5 Single Patient or Subject Viewer
This chapter describes the single patient or subject viewer of OHTR. It contains the following topics:
Viewing Records
The View Record tab helps you focus on the discreet medical history or genomic history of a particular patient. You can drill into the data details of a particular patient, and locate the attribute or data element that is pivotal as selection criteria for your cohort.
To view a patient record:
-
Enter the patient ID.
-
Select Source. By default, All records is selected, which lets search through all the patients available in the CDM database.
-
Select the clinical and genomic data you want to view for the patient.
-
Click Submit. The available patient information is displayed on the right.
-
To print the current screen, click Print at the bottom of the screen.
-
To save the data in an Excel file, click Export at the bottom of the relevant section.
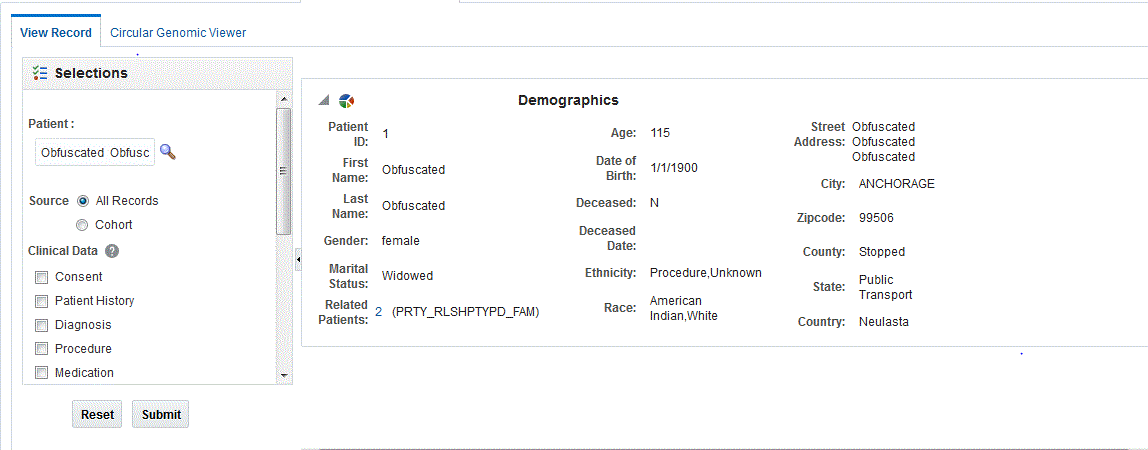
A patient or subject may have significant volumes of information and you can search for a particular attribute. Clicking the header of any column will sort the data within it.
-
Enter a value in the blank field at the top of any column.
-
Press Enter. The rows are filtered based on the attribute you have specified.
Note:
The attributes that display for the various categories align with the data terms and definitions that are described in the Cohort Query tab, where you select criteria for selecting patients or subjects.Navigating Through Selected Patients or Subjects
If you have a specified patient or subject count in the single patient or subject screen on the Cohort Query or Cohort List screen, you can navigate through all the selected patients or subjects clinical and genomic history by using the Previous and Next buttons.
Under the Patient field, the current Patient's ordinal number (ordinal position of the Patient record within the selection) and the total number of available Patients or Subjects (in the Cohort) will be displayed. For example, in the following figure it is (1 of 8) patients.
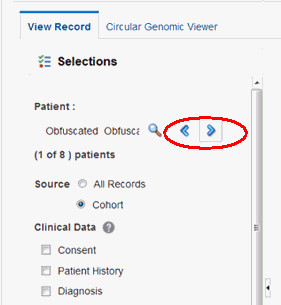
If you have navigated from the Cohort Timeline screen into the single patient or subject screen, then the total number of available patient or subject cohort will be the count of patients or subjects selected in the initial pool.
Genomic Data
Genomic data is displayed in four sections as follows:
Specimen with Genomic Results
Table 5-1 Specimens with Genomic Results
| Column Heading | Definition | Sample Value or Values |
|---|---|---|
|
Specimen Id |
Specimen belonging to the selected patient or subject |
HG00096 |
|
Specimen Vendor Id |
Specimen Vendor for that specimen |
Vcf |
|
Version Label |
Represents Assembly Version (DNA reference version against which this data was loaded) |
GRCh37(V68) |
|
Sequence Variants Results |
Whether the sample has sequence variants results |
Yes / No |
|
Copy Number Variation Results |
Whether the sample has copy number variants results |
Yes / No |
|
Single Channel Microarray Results |
Whether the sample has single channel results |
Yes / No |
|
Dual Channel Microarray Results |
Whether the sample has dual channel results |
Yes / No |
|
Rna-Seq Expression Results |
Whether the sample has rna sequencing results |
Yes / No |
Derived Files
| Column Heading | Definition | Sample Value or Values |
|---|---|---|
|
Filename |
Filename including path of the genomic file stored including path |
C:/John_specimen01.vcf |
|
File Size in MB |
Size of the File in MB |
Numeric, positive integer |
|
File URI |
URI of the file |
File://trc/abc.maf |
|
Alternate Filepath |
The FTP path of the file |
|
|
File Type, Version |
Type of file and Version |
Variant Call Format, 4.1 |
|
Result Type |
Type of result data in the file |
Sequencing, Copy Number Variation, Gene Expression (2-channel or single channel) |
|
Alignment Version (DNA Reference Version) |
Represents Assembly Version (DNA reference version against which this data was loaded) |
GRCh37(v68) |
|
Total Number of Specimen in File |
Total number of specimen that the file contains where not all specimen belong to the selected patient |
Numeric, positive integer |
|
Last Updated |
When record was last updated |
19-Mar-2012 |
File Lineage
Note:
If you have appropriate permissions, and if files are present in the middle tier accessible location, the path listed in the File Name fields for Genomic data are enabled to let click and download the files directly from OHTR.| Column Heading | Definition | Sample Value or Values |
|---|---|---|
|
Parent Filename |
Parent Filename including path of the genomic file stored including path |
C:/John_specimen01.BAM |
|
File Size in MB |
Size of the File in MB |
Numeric |
|
File URI |
URI of the file |
File://trc/abc.maf |
|
Alternate Filepath |
The FTP path of the file |
|
|
File Type, Version |
Type of file and Version |
Binary Alignment Map, 1.0 |
|
Alignment Version (DNA Reference Version) |
Represents Assembly Version (DNA reference version against which this data was loaded) |
GrCH37 |
|
Last Updated |
When record was last updated |
19-Mar-2012 |
|
Derived Child Files: File - Specimen Id, Vendor Id |
Information about Derived Results files that have their lineage based on the particular Low Level file. |
C:/John_specimen01.vcf - HG00096_1,HarvardLab1 |
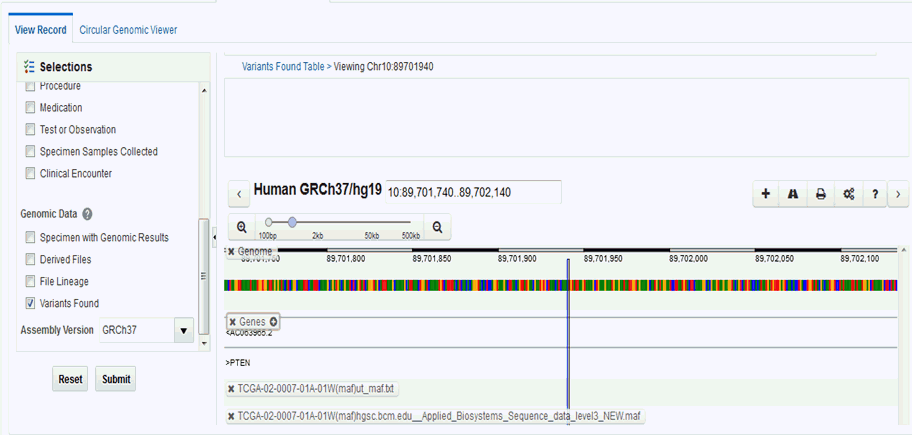
Variants Found
Selecting this option displays the different variants available for a patient. These variants are grouped and displayed in a hierarchical structure with the count of the variants displayed for each type of variants.
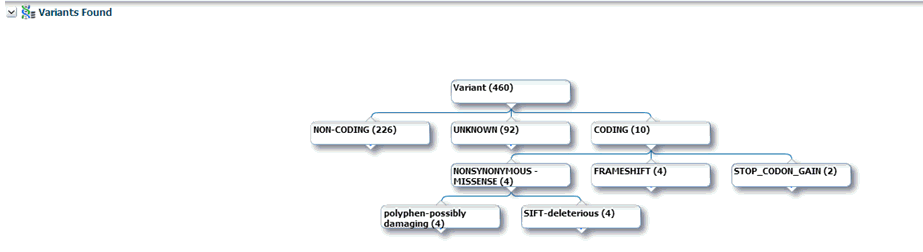
On selecting any of the nodes the details of the variants are displayed in a table as follows:
| Column Heading | Definition | Sample Value or Values |
|---|---|---|
|
Chromosome |
Chromosome location of the variant |
1 |
|
Position |
The position of the variant within the chromosome |
Numeric value |
|
Reference Allele |
The reference allele of the variant |
C |
|
Alternate Allele |
The alternate allele of the variant |
G |
|
Gene |
The gene containing the variant |
BID |
|
Transcript |
The transcript name |
ENSTXXX |
|
Variant Name |
The reference id of the variant |
rs111 |
|
Variant Type |
The type of variant |
Substitution |
|
Variant Status |
Status of the variant |
Known |
|
Protein Name |
||
|
SIFT Impact |
The SIFT impact of the variant |
Intolerant |
|
PolyPhen Impact |
The polyphen Impact of the variant |
Damaging |
|
Drug |
The related drug |
Clofazimine |
|
Associated Disease |
The disease associated to the variant |
Anaemia |
|
Histology |
||
|
Site |
||
|
Specimen Id |
The specimen containing the variant |
HG00096 |
|
Alignment Version |
Alignment version of a variant |
GRCh37 |
Selecting Assembly Version
By default, the last loaded assembly is displayed but this selection can be modified. The Genomic data is filtered out with this assembly version instead of the DNA reference version. One Assembly version can belong to multiple DNA reference versions.
Dalliance Browser
The Dalliance browser is a third party tool that displays a graphical representation of the variant, chromosome or gene range. The variant detail table comprises of hyperlinks for variant reference ID, gene name and chromosome position.
-
In the patient details section, scroll down to Variants.
-
The variant detail table comprises of hyperlinks for variant reference ID, gene name and chromosome position.
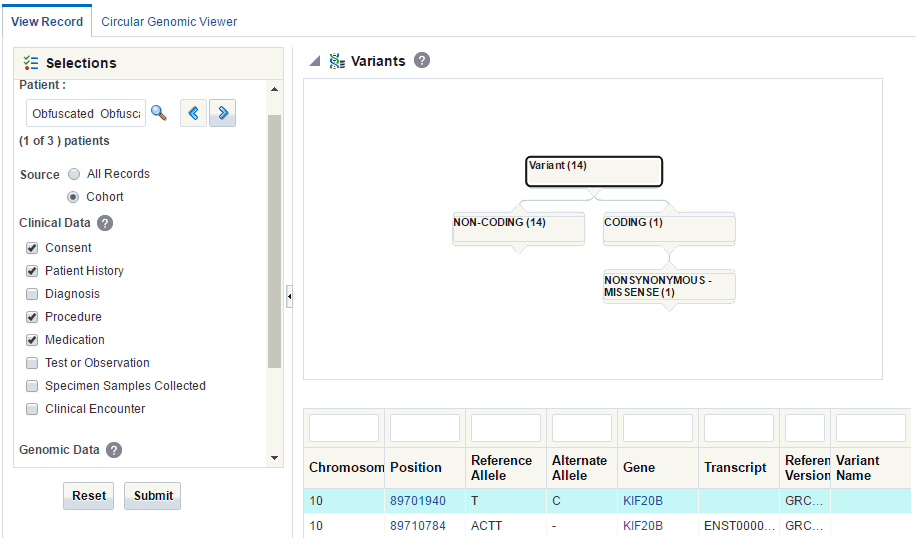
Description of the illustration ''trc102.gif''
-
Click any of these hyperlinks to navigate to the Dalliance browser where you can see the particular VCF tracks around a specific variant, a specific gene, or in an explicitly specified chromosomal range.
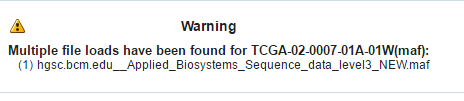
If a file is loaded multiple times for a particular specimen, then the following warning message is displayed. It lists all the multiple files names.
Description of the illustration ''trc126.gif''
Following are some configurations required to plot the gene track in the Dalliance browser. You can create your own DAS server and the corresponding entries should be added in the TRC.properties file. Following is an example of the required entries, to look up the Authority, University of California Santa Cruz (UCSC) name for the alignment and the public URLs for the Sequence and Genes tracks (reference tracks):
DALLIANCE.AUTHORITY_37=GRCh DALLIANCE.UCSC_NAME_37=hg19 DALLIANCE.SEQ_URL_HG18=http://www.derkholm.net:8080/das/hg18comp/ DALLIANCE.SEQ_URL_HG19=http://www.derkholm.net:8080/das/hg19comp/ DALLIANCE.GENES_URL_HG18=http://www. derkholm.net:8080/das/hsa_54_36p/ DALLIANCE.GENES_URL_HG19=http://www. derkholm.net:8080/das/hsa_59_37d/
While integrating Dalliance with OHTR, the server http://www. derkholm.net:8080/das/ has been used. However, this has been shut down.
These can be manually customized in TRC.properties. To add any new alignments, add all the above code to TRC.properties. Make sure that you research the reference for where they can be found. It is possible to use downloaded files instead of public DAS server(s) for reference, but the client should host these files on a web server.
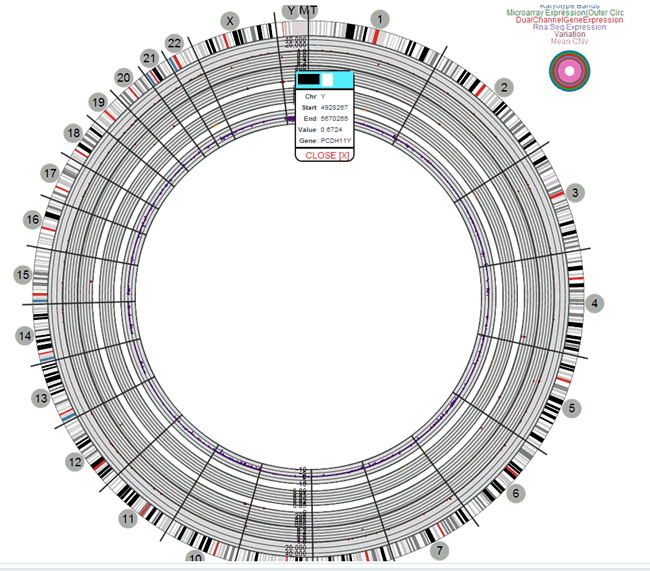
Circular Genomics Viewer
Circular genomic data viewer provides an interface for you to visualize the genomic data which includes variation, micro array expression, copy number variation, dual channel expression and RNA sequencing. The system uses the VisQuick tool, which is a Javascript library built specifically for genomic data visualization.
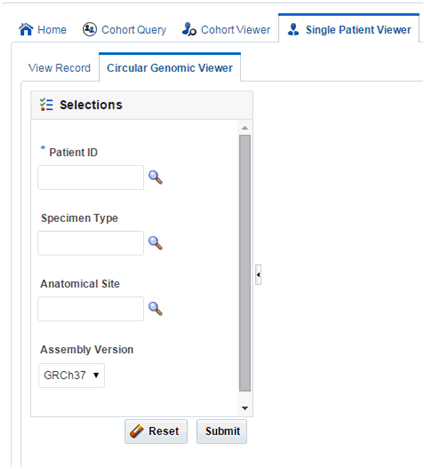
To view circular genomic data:
-
Select Patient ID or Subject ID. If the Patient or Subject ID has been selected in the View Record screen, it will be inherited here.
-
To add filter criteria, select Specimen Type and Anatomical Site. The DNA Reference Version selected is used to filter out the results and determine the cytoband to be used while rendering the circular genomic plot for any of the five data types.
-
Click Submit. Based on the filter criteria, the matching specimen in any of the five result types are displayed.
-
Select only one specimens of different result types to plot the graph. By default, the cytoband of chromosomes is also plotted which is the outer most ring of the circular plot.
-
Click Plot Graph. The circular genomic data graph is displayed.
Note:
For the circular viewer to run properly in IE9, turn off compatibility mode.
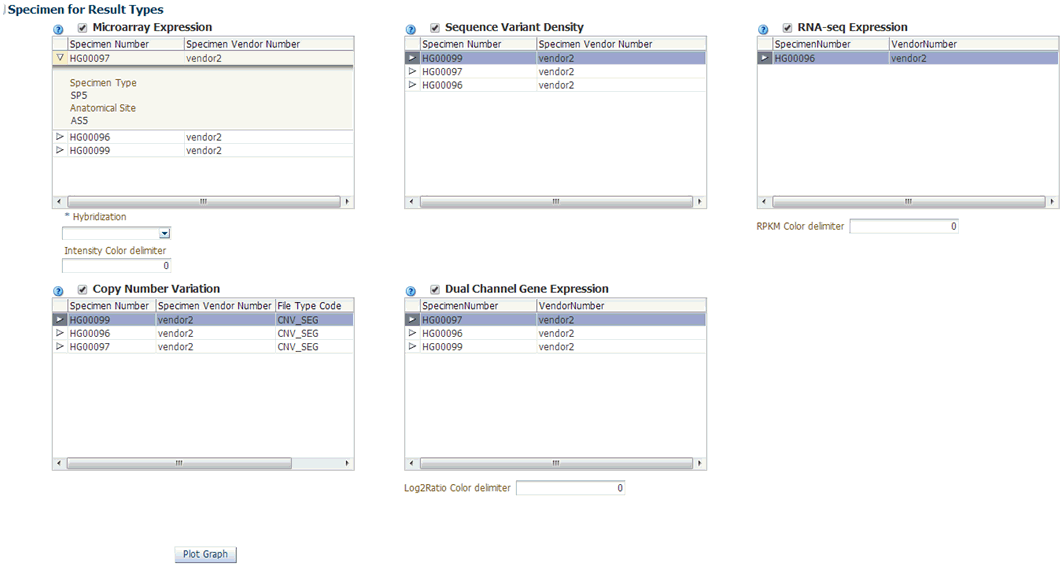
Selecting Data to Plot
Microarray Expression
The microarray expression panel displays all the specimens of the result type expression. You can select only one specimen at a time.
For the selected specimen from the panel, you can view the list of hybridizations available for that specimen. You can select maximum of two hybridizations from the multiple choice box, for which the plot is rendered.
The color delimiter value is used to render color to the data points in the plot based on the intensity value. If the intensity value is above the value defined in color delimiter then the data points have green color Otherwise, the points have red color.
Sequencing-Variant Density
This option enables you to plot the density of variants for every 100-kb region for a specimen. The sequence variant density panel displays all the specimens of the result type sequence variants. You can select only one specimen at a time.
RNA-Seq Expression
This panel displays all the specimens of result type RNA Sequencing.You can select only one specimen at a time.
The color delimiter value is used to render color to the data points in the plot based on the RPKM value. If the RPKM value is above the value defined in color delimiter then the data points have green color. Otherwise, the points have red color.
You can use the check boxes provided next to each of the result types to determine if the specimen selected will be plotted or not. You can select one or all of the result types for plotting of the graph. Once the specimens are selected, the circular genomic plot is displayed. The outermost circle in the plot is the Cytoband. The supported Cytoband versions are: hg-18 and hg-19.
Note:
To display the cytoband in the Circular Genomic plot we need an entry mapping each DNA Reference Version to the Cytoband in the TRC_LOOKUP_CODE table in the APP_schema.For each reference version we must insert a row with the following values:
-
CODE_TYPE : TRC_REFVERSION_CYTOBAND
-
CODE:cytobandHG19 (for HG19), cytobandHG18 (for HG18), or cytobandHG38 (for HG38)
-
CODE_NAME :Name of the loaded DNA reference version (for example, V69)
You can hover over the plot to get details such as:
-
Microarray Expression: Chromosome, Start Position, End Position, Value and Gene
-
Copy Number Variation: Chromosome, Start Position, End Position and Value
-
Sequence Variants: Chromosome, start position, end position, and value. The density value is calculated by: Total number of variants for 100kb region / 100000
-
Dual Channel Microarray Expression: Chromosome, Start Position, End Position, Value and Gene
-
Rna Seq Expression: Chromosome, Start Position, End Position, Value and Gene
Copy Number Variation
The panel displays all the specimens of the result type copy number variation. You can select only one specimen at a time.
The value for copy number variation depends on the CNV result type selected. If the selected specimen contains data from Genome_Wide_SNP_6 array, then the value for CNV is taken from segment mean stored in the database. If the selected specimen has data from complete genomics, then the value is calculated based on the calledPloidy value stored in the database. The value to be plotted is calculated using the following formula for CNV data from complete genomics:
log2(called_ploidy/expected_ploidy)
where,
expected_ploidy is 2 for chr1-22
expected_ploidy is 2 for chrX for females.
expected_ploidy is 1 for chrX outside the pseudo-autosomal region in males
expected_ploidy is 2 for chrX inside pseudo-autosomal region in males
The pseudo autosomal regions on chrX for 'NCBI build 37' as reported by Complete Genomic are 60000 - 2,699,519 and 154,931,043 - 155,260,559.
The pseudo autosomal regions on chrX for 'NCBI build 36' as reported by Complete Genomic are 0 -2,709,519 and 154,584,237 - 154,913,753.
For called_ploidy zero, there the log2 will be infinity, in such cases the final value is taken as -2.
Dual Channel Microarray Expression
This panel displays all the specimens of result type Dual Channel Microarray expression. You can select only one specimen at a time.
The color delimiter value is used to render color to the data points in the plot based on the Log2Ratio value. If the Log2Ratio value is above the value defined in color delimiter then the data points have green color. Otherwise, the points have red color.