3 View and maintain cohorts
In this chapter, you will find out how to:
View cohort lists
-
On the Home page, on the left, click the Cohort Viewer icon (
 ).
). -
On the left side, in the Patients section, choose your option:
-
active query: to view the cohort list for a patient or subject query that is currently running
-
library query: to view the cohort list for a patient or subject query from the library
-
list: to view the cohort list for a saved patient or subject list
-
ad-hoc query: to view lists for specific patients or subjects you want to examine
-
-
Click Submit.
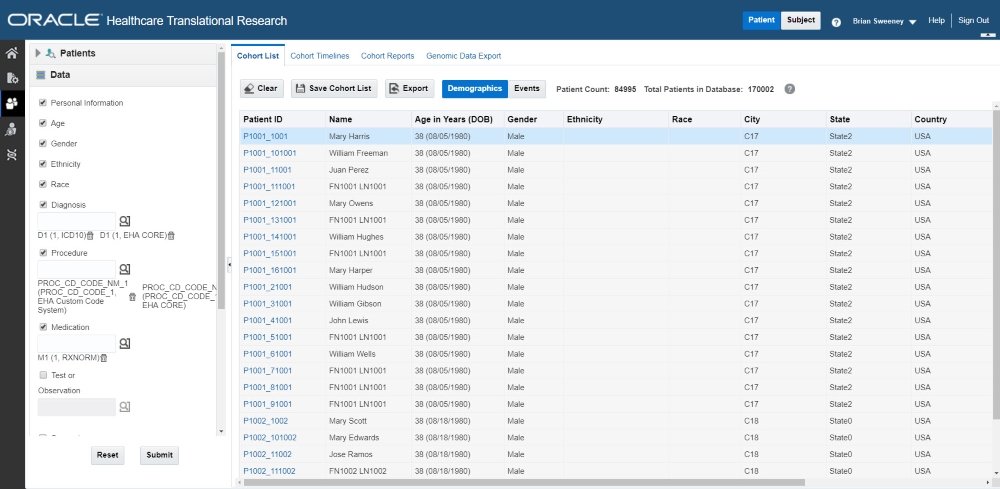
Use the Data pane on the left to select and filter the data you want to view for the cohort list. For details on filtering data, see Filter data in cohort lists.
After running the query, the patient or subject count is displayed above the table, and you can review the data of those patients or subjects.
Note:
Patient or subject count can also be viewed in the Cohort Query screen as well. For details, see View Patient or Subject Count and data.Filter data in cohort lists
-
On the left, below the Patients section, click Data.
-
Select or deselect any of the information that you want included in the cohort lists.
-
Either click the radio button for Include criteria used in a query or set your custom criteria.
Note:
The Include criteria used in a query can only be used for active queries.This option filters clinical records in cohort lists and cohort timelines only on the basis of the primary coded attributes from the Cohort Query screen. For example, it only uses Diagnosis Codes, Procedure Codes or Medication codes.
If the inclusion criteria of the active query is not based on any data topics supported in the cohort lists, a message is displayed to select at least one supported data element.
To set custom criteria:
-
Select the criteria.
-
Enter the criteria or click the search icon (
 ).
). -
In the new window, enter all required details and click Search.
-
Select the elements that you want to use from the left side and move them to the right side using the arrow (
 ).
). -
Click Submit.
-
-
Click Submit.
To remove data, clear the appropriate boxes and click Submit again.
To export the list to a Microsoft Excel sheet, click Export ( ![]() ), along the top of the table. Keep in mind that additional information, such as address and email are only visible in the exported XLS file.
), along the top of the table. Keep in mind that additional information, such as address and email are only visible in the exported XLS file.
To save a cohort list, click the Save button ( ![]() ). You can rename the list, or save it under an existing name. All cohort lists are saved as Private.
). You can rename the list, or save it under an existing name. All cohort lists are saved as Private.
To switch between Demographic and Events data, use these buttons above the table:
Tip:
To display the date in the dd/mm/yyyy format, use the formatting option in Microsoft Excel.Display reference range values
Cohort lists can display reference range values, if available, along with numeric results of observation.
-
In the Cohort Viewer screen, on the left, click and the Data section.
-
Click the radio button for Test or Observation.
-
Enter the value you want to search for.
-
Click Submit.
Where available, the reference high and reference low range values are displayed along with numeric results in the cohort list.
View cohort timelines
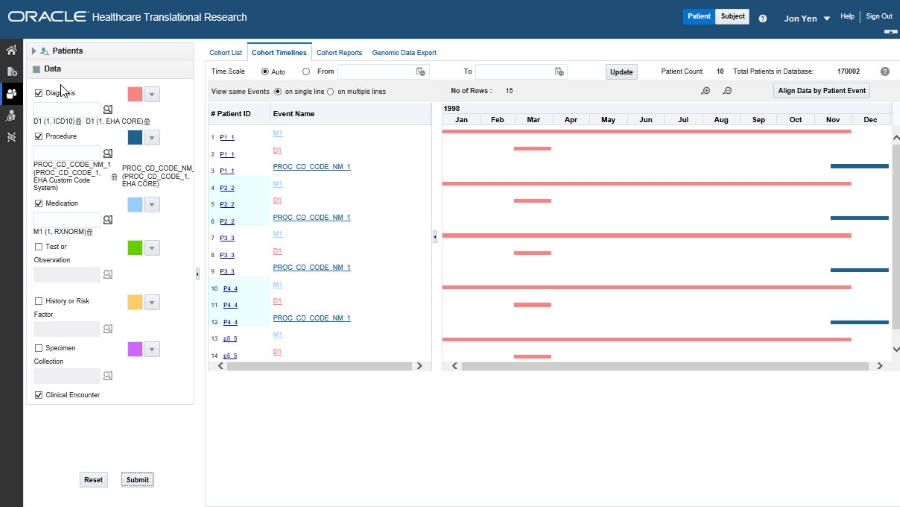
Select patients or subjects
This step is similar to how you select patients or subjects for the Cohort List.
-
Along the top, click the Cohort Timelines tab.
-
On the left, expand Patients or Subjects to select the source of your patients' or subjects' IDs from an active query, library query, list or ad-hoc query.
-
Click Make initial pool.
All patients or subjects from the selected query are added to the initial pool.
-
Select up to 20 patients or subjects and click the arrow (
 ) to move them from the initial pool to the display list.
) to move them from the initial pool to the display list.The data displayed in the cohort timeline is of these 20 patients or subjects.
WARNING:
Don't click Submit yet! Wait until the next section.
Filter data in cohort timelines
Tip:
Each data topic has a distinct color associated with it, so that you can visually separate data for the same patient or subject. You can change the assigned colors by using the drop-down arrow next to each color.-
On the left, click the Data section.
-
Change the colors of the data criteria, if needed.
-
Either click the radio button for Include criteria used in a query or set your custom criteria.
Note:
The Include criteria used in a query can only be used for active queries.This option filters clinical records in cohort lists and cohort timelines only on the basis of the primary coded attributes from the Cohort Query screen. For example, it only uses Diagnosis Codes, Procedure Codes or Medication codes.
If the inclusion criteria of the active query is not based on any data topics supported in the cohort lists, a message is displayed to select at least one supported data element.
To set custom criteria:
-
Select the criteria.
-
Click the search icon (
). -
In the newly opened window, enter all required details and click Search.
-
Select the elements that you want to use from the left side and move them to the right side using the arrow (
). -
Click Submit.
-
-
Now that you have your data set, click Submit again.
Display patient or subject data
The subject and patient data display is divided into two sections.
-
The left side has attributes about the selected patients/ subjects: Patient/ Subject ID and Event Name.
-
To see demographic data for a Patient or Subject, hover over their ID.
-
The right side shows the colored layers of the selected clinical data, in a timeline view.
-
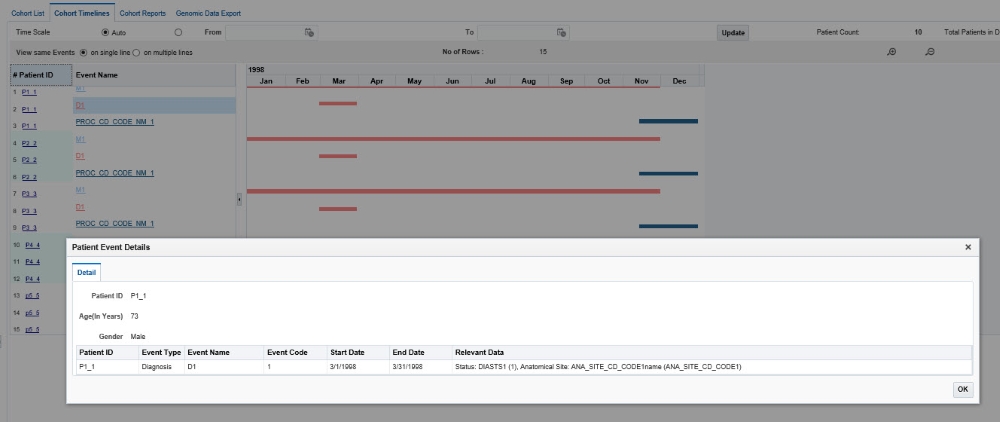
To see more details of a patient or subject and their medical records, click on an Event link from the left side table. A pop-up appears with more data on the event.
-
To navigate to the Single Patient Viewer, and see details of a specific subject or patient, click the Subject or Patient ID.
-
To view data for a specific date range, along the top, use the Time Scale options:
-
To view data for certain date range, enter the From and to dates and click Update.
-
To view data without a specific time reference, select the Auto option.
-
-
Since the Test or Observation event has only one date event, the user interface uses dummy End Date values that are 1 day later than the corresponding Start Date values to render the graph. The Detail table also displays these dummy End Date values.
-
Events are not displayed on the left-hand table or on the timeline in the following conditions:
-
When an event's Start Date is null.
-
When an event has only one Date attribute (for example, Observation has only Observation Date) and the attribute's value is null.
-
-
If an event's End Date is null, then the System Date is used for displaying the event.
Select the Timeline Mode
To view events in different ways, go to View same Events along the top and select one of the radio buttons:
This is the default mode of displaying data in a timeline.
In this mode, all instances of a repeating event are displayed on the same line and separate events are displayed on separate lines.
In this mode, all of instances of events are displayed on different lines, even if the events are repeating.
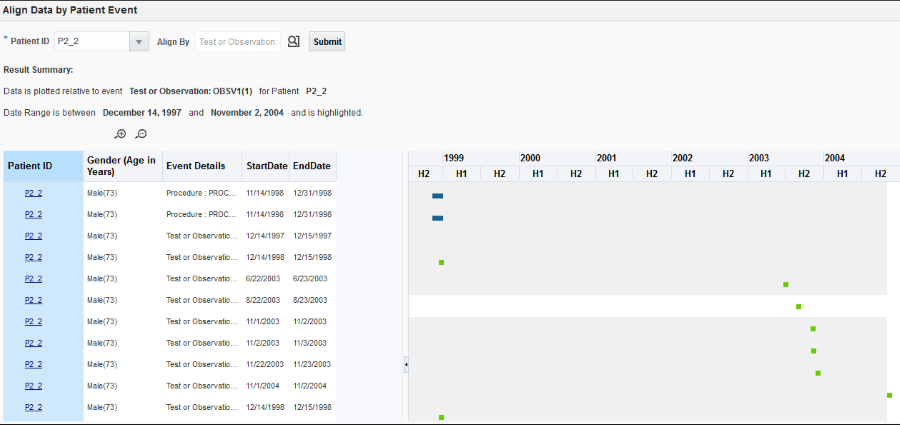
Align data by Patient or Subject Event
-
Click the Align Data by Patient or Subject Event button (
 ).
). -
In the new window, select a Patient or Subject ID from the drop-down list.
-
Click the search icon (
) next to the Align by field, and select the option you want to use.Note:
When you select a particular diagnosis or medication code to align patients, all event codes at different hierarchical nodes are shown for the selected event code. -
Click Submit.
View cohort reports
The Cohort Reports tab lets you view various cohort reports.
Note:
In the Active Query mode, if no filters have been selected in Cohort Query, then the reports display data for only the first 10,000 patients or subjects. This limit is configurable and can be changed using the DEFAULT_ACTIVE_QUERY_LIMIT property in the TRC.properties file.However, irrespective of the patient or subject selection option, if the count exceeds the value specified in GENOMIC.REPORTS_MAX_PATIENT_COUNT of TRC.properties file, a warning message is displayed. You can either continue to plot with a large number of patients or subjects (might impact performance) or change the selected cohort to a smaller number.
View demographic reports
-
Along the top, click the Cohort Reports tab.
-
On the left, click the Patients or Subjects section, and select a source for your reports, if needed.
-
Click Demographic Reports.
-
Select the check boxes for the data you want displayed in your list.
-
Click Submit.
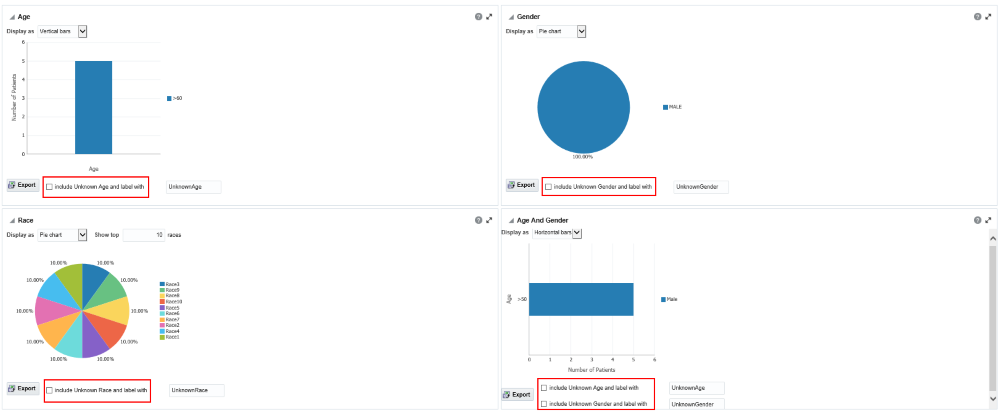
Handle unknown data
If any missing or unknown data is present, a check box is provided at the bottom of each graph to include missing or unknown data.
For the Age, Gender and Race graphs, if the check box is selected, the graph is refreshed with the unknown data. In this case, unknown data refers to age, gender or race details that are not defined for a particular patient.
For the Age and Gender graph, the unknown age for different genders is combined into one group and the known age is in another group. Both groups are shown in the graph.
View clinical reports
-
Along the top, click the Cohort Reports tab.
-
On the left, click the Patients or Subjects section, and select a source for your reports, if needed.
-
Click Clinical Reports.
-
Select the Specimen Available check box.
-
Click Submit.
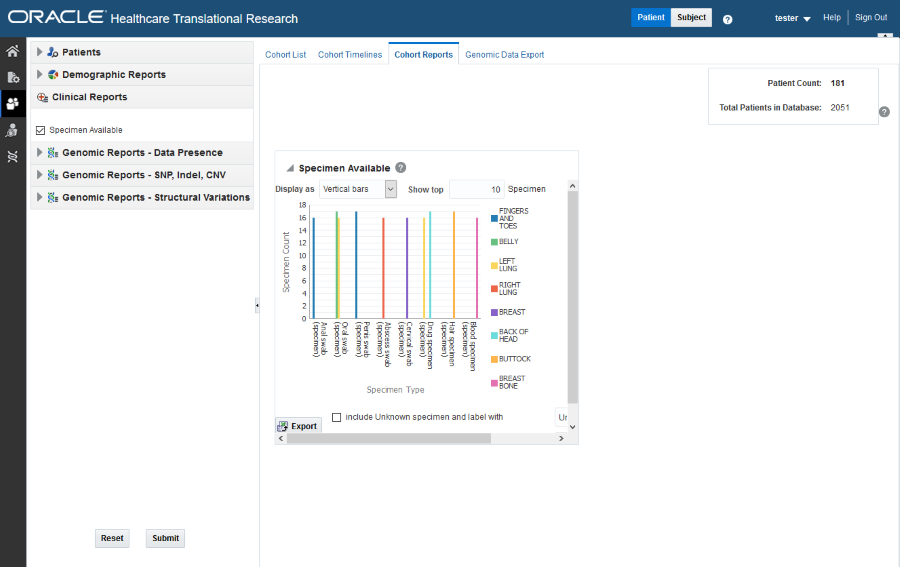
Handle Unknown Data
If any missing or unknown data is present, a check box is provided at the bottom of each graph to include missing or unknown data.
If Include Unknown Specimen and Label With or Include Unknown Anatomical site and Label With are selected, the graph is refreshed with the unknown data. In this case, unknown data refers to the specimen and anatomical site data that is not defined for a particular patient.
View genomic reports
View genomic data presence reports
-
Along the top, click the Cohort Reports tab.
-
On the left, click the Patients or Subjects section, and select a source for your reports, if needed.
Note:
There is no upper limit on the number of patients or subjects to be selected, however the performance slows down as more and more patients are selected. -
Click Genomic Reports - Data Presence.
-
Select the Genomic Data Presence check box.
An optional Specimen Type input field appears to allow for filtering the initial cohort on specimen type.
-
Click Submit.
Tip:
To change the display type after generating the report, select the display option from the Display as drop-down list.To view a breakup of present genomic data by data type, click View distribution of genomic data.
To export the graphical plot type as a PNG image, and the table type as an Excel spreadsheet, click the Export button( ![]() ) for every graphic.
) for every graphic.
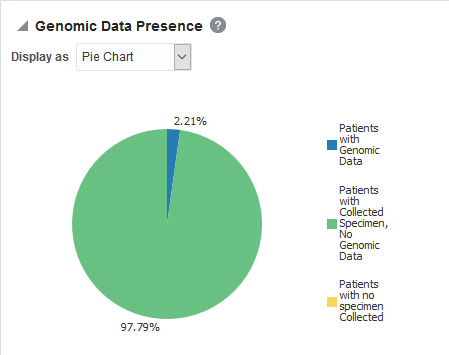
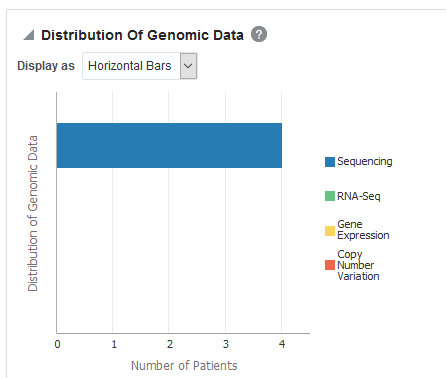
View Genomic Reports - SNP, Indel, CNV
Note:
Genomic Reports SNP, Indel and CNV display the available SNP Indel genomic reports based on the selected cohort of patients or subjects.Mutated Gene Frequency and Gene Expression
To view these reports:
-
Along the top, click the Cohort Reports tab.
-
On the left, click the Patients or Subjects section, and select a source for your reports, if needed.
Note:
There is no upper limit on the number of patients or subjects to be selected. However the performance may slow down as the number of patients or subjects increases. -
Click Genomic Report - SNP, Indel, CNV.
-
Select the radio button for Gene Level Reports.
-
Select Mutated Gene Frequency and Gene Expression.
Selecting Include All Specimens includes specimens without genomic data. Not selecting this option generates results with genomic data only.
-
Enter or search for at least one gene using one of the three search fields: Ad-hoc List, Pathway or Gene Set.
-
Expand the Additional Parameters section to add a Specimen Type or an Anatomical Site.
-
Click Submit.
You can display the results as horizontal bars, vertical bars, or as a table. To export the bar results into PDF or the table results into Excel, click the Export button ( ![]() ).
).
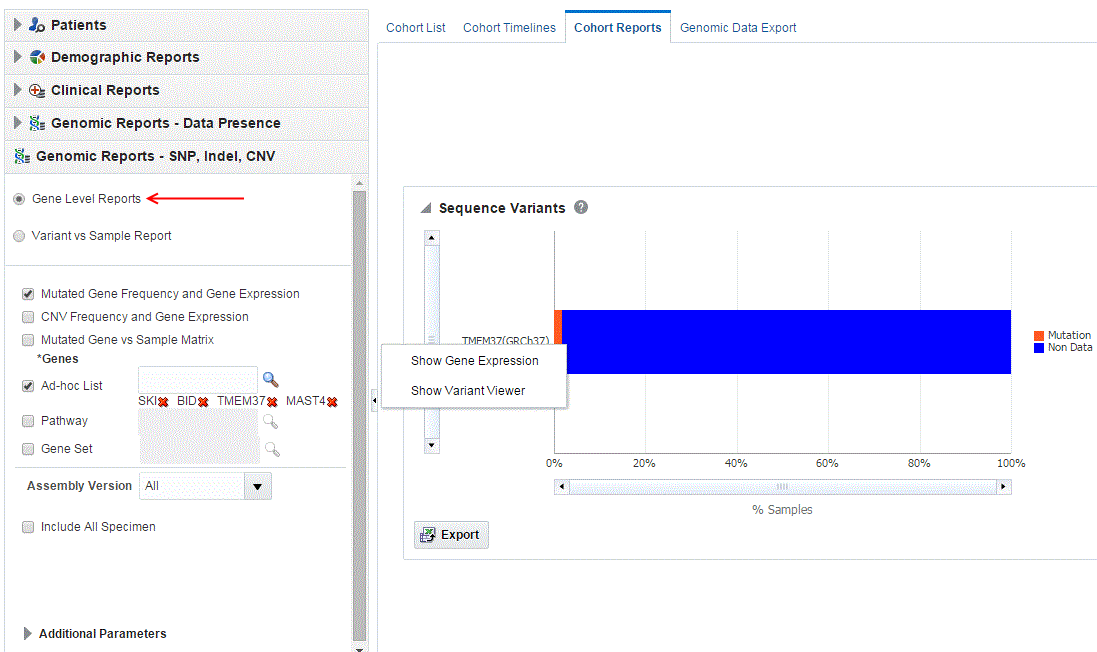
About the Mutated Gene Frequency and Gene Expression
Each bar represents a specimen, which is grouped patient-wise and in the order of the specimen collection date. The Sequence Variants and CNV information for each specimen is displayed. The percentage of specimens with mutation on the gene is also mentioned. Export functionality is provided at each specimen level.
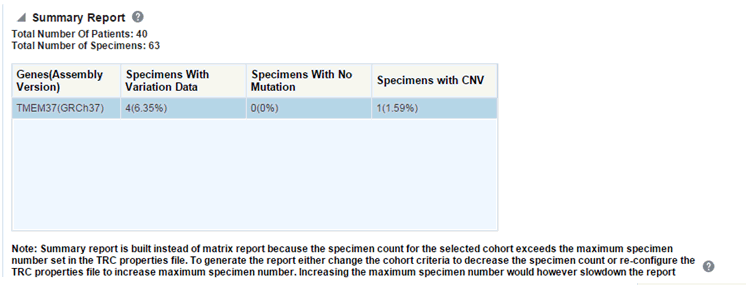
There is a limit to the number of specimens that can be seen in the gene matrix plot. The default value of this parameter is 1000. If the number of specimens in the cohort used for this analysis is greater than the specified value, then the following warning message is displayed:
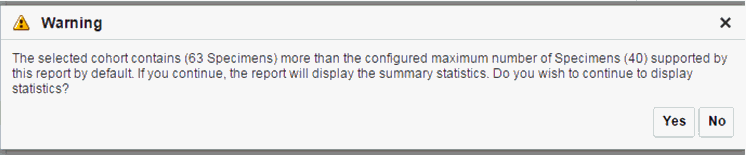
If you continue from this warning message, the report displays the summary statistics instead of the matrix plot. You may have to decrease the cohort size based on some criteria like specimen type or anatomical site or otherwise use cohort query to view the matrix plot. Alternatively, you can also change the default value of MAX_SPEC_REPORT parameter to a desired value and rebuild the plot. However, rebuilding maybe affect the performance of the plot generation.
Copy Number Variation Frequency and Gene Expression
To view these reports:
-
Along the top, click the Cohort Reports tab.
-
On the left, click the Patients or Subjects section, and select a source for your reports, if needed.
Note:
There is no upper limit on the number of patients or subjects to be selected. However the performance may slow down as the number of patients or subjects increases. -
Click Genomic Reports - SNP, Indel, CNV.
-
Select Gene Level Reports.
-
Select CNV Frequency and Gene Expression.
Selecting Include All Specimens includes specimens without genomic data. Not selecting this option generates results with genomic data only.
-
Enter or search for at least one gene using one of the three search fields: Ad-hoc List, Pathway or Gene Set.
-
Expand the Additional Parameters section to add a Specimen Type or an Anatomical Site.
-
Click Submit.
Note:
When opening the Excel file, you may receive a warning from Excel stating that the file is in a different format than specified by the file extension. This warning can be safely ignored. For more information, refer tohttp://docs.oracle.com/cd/E23943_01/web.1111/b31973/af_table.htm#autoId34.
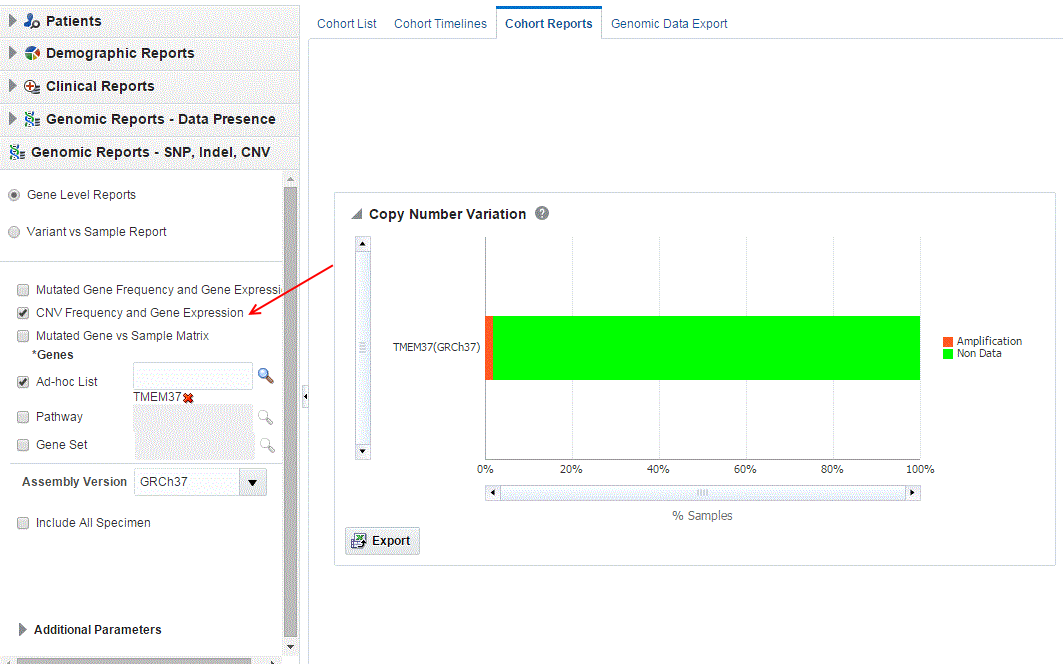
To export the bar results into or table results into Excel by clicking Export ( ![]() ).
).
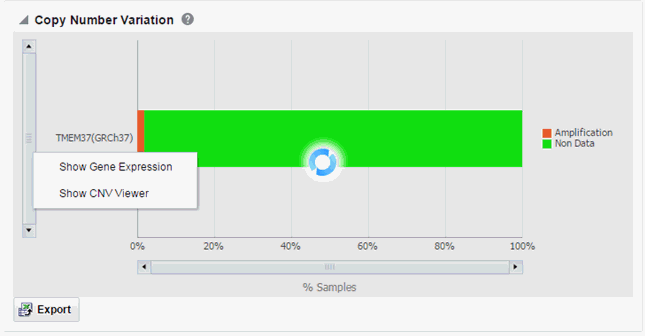
Select Show Gene Expression to display the Gene Expression details for Single Channel or Dual Channel. In the Single Channel Expression, you must provide hybridization details.
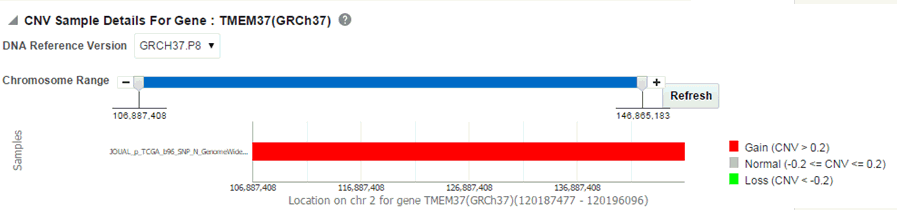
Click Show CNV Viewer to display necessary information about all CNV variants that are present in that gene in the samples belonging to the selected cohort of subjects or patients.
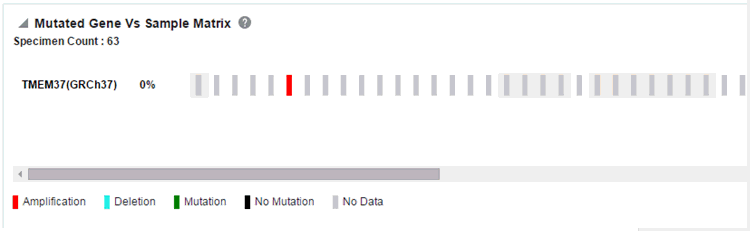
Mutated Gene vs Sample Matrix
To view this plot:
-
Along the top, click the Cohort Reports tab.
-
On the left, click the Patients or Subjects section, and select a source for your reports, if needed.
Note:
There is no upper limit on the number of patients or subjects to be selected. However the performance may slow down as the number of patients or subjects increases. -
Click Genomic Reports - SNP, Indel, CNV.
-
Select Gene Level Reports.
-
Select Mutated Gene vs Sample Matrix.
-
Enter or search for at least one gene using one of the three search fields: Ad-hoc List, Pathway or Gene Set.
Selecting Include All Specimens includes specimens without genomic data. Not selecting this option generates results with genomic data only.
-
Expand the Additional Parameters section to add a Specimen Type or an Anatomical Site.
-
Click Submit.
Note:
When opening the Excel file, you may receive a warning from Excel stating that the file is in a different format than specified by the file extension. This warning can be safely ignored. For more information, refer tohttp://docs.oracle.com/cd/E23943_01/web.1111/b31973/af_table.htm#autoId34.
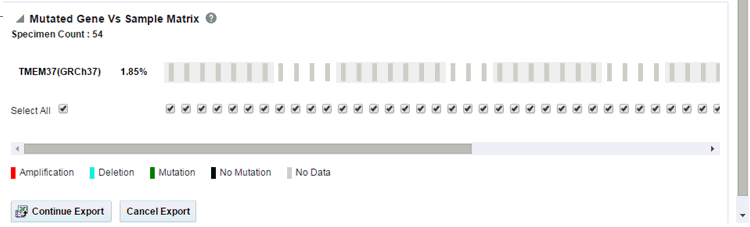
Variant vs Sample Reports
To generate the plot:
-
Along the top, click the Cohort Reports tab.
-
On the left, click the Patients or Subjects section, and select a source for your reports, if needed.
Note:
There is no upper limit on the number of patients or subjects to be selected. However the performance may slow down as the number of patients or subjects increases. -
Click Genomic Reports - SNP, Indel, CNV, and select Variant vs Sample Report.
-
Enter or search for a Variant ID.
Selecting Include All Specimens includes specimens without genomic data. Not selecting this option generates results with genomic data only.
-
Expand the Additional Parameters section to add a Specimen Type or an Anatomical Site.
-
Click Submit.


Note:
When opening the Excel file, you may receive a warning from Excel stating that the file is in a different format than specified by the file extension. This warning can be safely ignored. For more information, refer tohttp://docs.oracle.com/cd/E23943_01/web.1111/b31973/af_table.htm#autoId34.Structural Variations
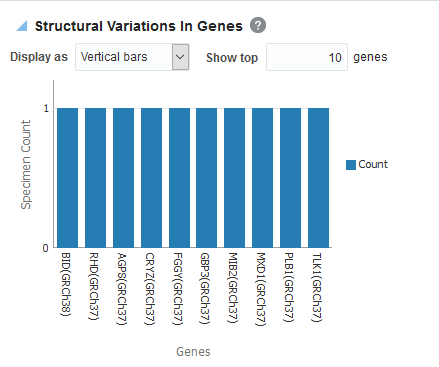
Structural Variations in Genes
To view the structural variations in genes:
-
Along the top, the click Cohort Reports tab.
-
On the left, click the Patients or Subjects section, and select a source for your reports, if needed.
Note:
There is no upper limit on the number of patients or subjects to be selected. However the performance may slow down as the number of patients or subjects increases. -
Click Genomic Reports - Structural Variations and select Structural Variations in Genes.
-
Expand the Additional Parameters section to add a Specimen Type or an Anatomical Site.
-
Click Submit. By default, the top 10 genes are shown and you can increase or decrease the number of displayed genes.
To export the bar results in PDF or table results in Excel, click Export (
 ).
).
Note:
When opening the Excel file, you may receive a warning from Excel stating that the file is in a different format than specified by the file extension. This warning can be safely ignored. For more information, refer tohttp://docs.oracle.com/cd/E23943_01/web.1111/b31973/af_table.htm#autoId34.Structural Variations in Gene Pairs
To generate a Structural Variations in Gene Pairs report in Subject context:
-
Along the top, click the Cohort Reports tab.
-
On the left, click the Patients or Subjects section, and select a source for your reports, if needed.
Note:
There is no upper limit on the number of patients or subjects to be selected. However the performance may slow down as the number of patients or subjects increases. -
Click Genomic Reports - Structural Variations and select Structural Variations in Gene Pairs.
-
Expand the Additional Parameters section to add a Specimen Type or an Anatomical Site.
-
Click Submit. By default, top 10 gene pairs are shown and you can change the default number of bars shown.
To export the bar results in PDF or the table results in Excel, click Export (
).Note:
When opening the Excel file, you may receive a warning from Excel stating that the file is in a different format than specified by the file extension. This warning can be safely ignored. For more information, refer tohttp://docs.oracle.com/cd/E23943_01/web.1111/b31973/af_table.htm#autoId34.
Export genomic data
The Genomic Data Export page is used to export the genomic data for filtered patients or subjects based on Study, Specimen type and Anatomical Site in a specific file format.
Note:
Currently, exporting variation data from sequencing platform, single and double channel gene expression and Copy Number Variation data in VCF, SEG, RES and GCT file formats are supported. These formats are supported by the IGV browser.Select results to export
Currently only one version of data can be exported at a time. To select results:
-
On the left, click the Patients or Subjects section, and select a source for your results.
-
Select the Assembly Version from the drop-down list.
-
Enter a Specimen Type or search for one using the search icon (
). -
Enter an Anatomical Site or search for one using the search icon (
).
Set the location
You can export genomic data using three different methods:
-
Click the radio button for In Genes from and select one or all options:
- Pathway so you can select one or more pathways.
- Gene Set, so you can use the user-defined collection of genes.
- Ad-hoc List so you can select one or more genes.
-
Click the radio button for At Genomic Position and enter a specific chromosome region.
The following chromosome region formats are supported:
CHR15:10000-200000: Considers region between 10000 to 200000 in chromosome 15. CHR15:1,200,000+5000 - Considers 5000 bases upstream from 1,200,000 position in chromosome 15. CHR15 - Considers whole of the chromosome 15. CHR15:1000 - Considers 1000th nucleotide position of chromosome 15.
-
Click the radio button for All Data to export the complete genomic data of patients or subjects.
Select file type
Select one or all of the available file types:
-
Mutation - VCF
-
Copy Number Variation - SEG
-
Microarray Expression - RES
-
Microarray Expression Dual Channel - GCT
Note:
For the At Genomic Position location selection, the Gene Expression - RES and Gene Expression Dual Channel options are disabled.Mutation - VCF
This option exports the sequencing variation data for the selected patients or subjects for either the selected genes, pathway, gene set or for a given chromosome region as selected in the previous option. VCF supports multiple specimens' data in a single file.
The metadata header gives the following information that differs based on the search criteria:
-
##fileformat=VCFv4.1
-
##fileDate: Date and time of the VCF file generated.
-
##source=Oracle Healthcare Omics (OHO, formerly known as Omics Data Bank)
-
##Total Number of patients included in this VCF file
-
##Total Number of samples included in this VCF file
-
7. ##INFO=<ID=NS, Number=1, Type=Integer, Description=Number of Samples With Data>
-
##FORMAT=<ID=GT,Number=1,Type=String,Description=Genotype>
-
##FORMAT=<ID=GQ, Number=1, Type=Integer, Description=Genotype Quality>
-
##FORMAT=<ID=GQVAF, Number=2, Type=Integer, Description=Genotype_quality_X>
-
##FORMAT=<ID=DP, Number=1, Type=Integer, Description=Read Depth>
-
##FORMAT=<ID=AD,Number=.,Type=Integer,Description=Allelic depths for the ref and alt alleles in the order listed >
-
##FORMAT=<ID=HQ, Number=2, Type=Integer, Description=Haplotype Quality>
-
##FORMAT=<ID=BQ,Number=.,Type=Integer,Description=Average base quality >
-
##FORMAT=<ID=MQ,Number=.,Type=Integer,Description=Average mapping quality >
-
##FORMAT=<ID=SS,Number=1,Type=Integer,Description=Variant status relative to non-adjacent Normal,0=wildtype,1=germline,2=somatic,3=LOH,4=post-transcriptional modification,5=unknown>
-
##FORMAT=<ID=SSC,Number=1,Type=Integer,Description=Somatic Score>
The following data types are imported to VCF file:
| Data Type | Description |
|---|---|
| CHROM | chromosome |
| POS | position of the variation |
| ID | dbSNP ID or COSMIC ID associated with a variant |
| REF | reference allele |
| ALT | variant alleles |
| QUAL | not populated. Will have '.' specified in this column. |
| FILTER | is populated as PASS |
| INFO | Not populated. Will have '.' specified in this column. |
| FORMAT:GT | genotypic data for each specimen. |
| FORMAT:GQ | genotype quality. If not value available in DB, then '.' is specified in the file. |
| FORMAT:GQX | mapped to GENOTYPE_QUALITY_X column |
| FORMAT:DP | this stores the TotalReadCount for a specific variant |
| FORMAT:AD | this stores the reference read count and Allele read count for a specific variant. |
| FORMAT:HQ | not populated as of now. Will have '.' specified in this column. |
| FORMAT:FT | this stores GENOTYPE_FILTER column value |
| FORMAT:BQ | stores the RMS base quality |
| FORMAT:MQ | stores the RMS mapping quality |
| FORMAT:SS | stores the somatic status |
| FORMAT:SSC | stores the somatic status score value |
| Flex field format | If any custom formats are available, they are also included in the export. |
1000 Genomes VCF 4.1 conventions are followed while exporting variation data, however certain data types, which are non-standard, like BQ and MQ, may differ in convention for some customers since there is no standard way to represent them.
Handle Non-variant and No-call Data
If NON_VARIANT and (or) NOCALL records exist for any given position, the zygosity is checked to determine if the information format from these tables is used.
Note:
For het-ref or half zygosity values, these other format fields are compared with the existing SEQUENCING information. This information is then used with zygosity to create the format string.The NON_VARIANT data allows for GQ, GQX, MQ, BQ and the first reference read count of AD. The NOCALL data allows for all format fields to be compared. Both NON_VARIANT and NOCALL do not support exporting flex fields. The GT value of the format string reflects the stored zygosity as follows:
| Zygosity | FORMAT string GT:GQ:GQX:BQ:MQ:AD:DP |
|---|---|
| het-ref | 1/0:99:98:38:45:20:10,10 |
| Half | 1/.:99:98:34,34:45,45:20:10,5 |
| Het-alt | 1/2:99:98:43,44:56,67:20:0,10,10 |
| Hom | 1/1:99:98:34,34:45,45:20:0,19 |
If there are no result records for any specimen, the export displays "." with no other information for the format.
Handle Duplicate Genetic Information Exports
There could be cases where users reload genetic information multiple times for the same specimen. This may create ambiguous values for the different fields that exist in the VCF export file. The export code deals with such ambiguous numerical values that represent the quality (that is, GQ, GQX, AD, BQ, MQ). This code now computes minimum values and ensure that the value of least confidence is reported. There could be more complex cases, for instance, if there are 2 different alleles for the same position belonging to the same specimen, or variants with same position for same specimen with different zygosity. The export code uses MIN functions on all values including all the text fields. This allows for VCF export to create a valid file that can be loaded into genome browsers.
Alternatively, you can choose not to consider data from a specific specimen or a specific file using following methods:
-
Using DELETE_FLG - A user may load results for a specimen more than once that can completely contradict previous results. Users can set the DELETE_FLG as 'Y' on W_EHA_RSLT_SPECIMEN and (or) W_EHA_SPEC_PATIENT or W_EHA_SPEC_SUBJECT to have previous loads excluded, and then reload the correct result files. When the user now exports the data, only the latest loaded specimen data is considered for export.
-
Using FILE_URI - Oracle recommends using this method since you need not reload the data again as opposed to the above method. When there are multiple files loaded with contradicting data for the same specimen, user can set some files as obsolete by changing the W_EHA_FILE_LOAD.FILE_WID column. For example, if you have loaded the same specimen data 3 times and would like to consider the latest file loaded for export, then you must first identify the latest FILE_WID from W_EHA_FILE_LOAD table. Then change the FILE_WID of two old files in W_EHA_FILE_LOAD table to the latest FILE_WID. Now, all the three records belonging to the three file loads contain same FILE_WID, which represents the latest file load and only the latest file export data is exported.
Allele depth values represented under the AD data type are in the order of the alleles represented in the GT. Refer to the following table with examples:
| ALT | FORMAT | SAMPLE1 | |
|---|---|---|---|
| G,C,T | GT:AD | 1/2:0,4,6 | 0 represents reference_read_count
4 represents allele_read_count of 'G' 6 represents allele_read_count of 'C' |
| G,T | GT:AD | 2/2:0,4 | 0 represents reference_read_count
4 represents allele_read_count of 'T' |
| G,T | GT:AD | 1/0:10,5 | 10 represents reference_read_count
5 represents allele_read_count of 'G' |
Copy Number Variation - SEG
The copy number variation data is exported in SEG format. Currently, CNV data from any array based system like Affymetrix Genome Wide SNP 6 array whose data is in SEG format while loading in OHO is supported. The main requirement for exporting CNV data is to have the SEG_MEAN value in the CNV table of OHO.
For exporting data that is not loaded from SEG files, for example, data from CGI CNV files or any other source of CNV data, users have to create their own loader. The loader is expected to calculate the SEG_MEAN value since this value is most important for export.
-
ID: specimen ID of the reported CNV segment
-
chrom: chromosome name
-
loc.start: start position of the CNV segment
-
loc.end: end position of the CNV segment
-
num.mark: for array based CNV data, this stores the number of probes details
-
seg.mean: this stores the segment mean value from SEG_MEAN column in CNV table.
Gene Expression - RES
RES is one of the gene expression formats supported by IGV browser. Currently, only microarray gene expression data is exported to this format. Following data types are imported to RES format:
-
Description: hugo name of a specific probe
-
Accession: probe ID
-
Intensity: intensity value of the associated probe
-
Call: call of the associated probe
Gene Expression Dual Channel - GCT
GCT is one of the gene expression formats supported by IGV browser. Currently, only AgilentG4502A platform microarray gene expression data is exported to this format. Following data types are imported to GCT format:
-
Description: Gene symbol of a specific probe
-
Accession: probe ID
-
Intensity: intensity value of the associated probe
-
Call: call of the associated probe
Note:
The GCT file takes its gene symbol for the probe from the 2-channel composite element of ADF file. This is input into the ADF composite table in OHO. This value may not match with HUGO name in certain cases as OHTR associates 2-channel records in the result table that has partial (which includes a flanking region set by the user) genomic coordinate. The coordinate overlaps between composite elements and gene segments in the reference. This may also result in some cases in more than one unique gene in the reference mapping to a gene composite element.Specify the export options
Export data in three ways:
-
Select the option to download last loaded file(s)
-
Immediately, which is the default option
-
Schedule
After selecting the Export option, click Submit to finalize the genomic data export. The data is generated and a separate link is provided in the bottom panel for each result type.
The Immediately option gives you the file link on the same screen and you can click to download it immediately. The link provided has a specific naming convention: <file type>_OHO_<date:MM-DD-YYYY>_<time:HH24*-MI-SS>.<file_type_extension>. For example, RES_OHO_09-14-2014_04-26.res. A short description of the file stating data type and advice on the expected count of features is displayed below the created link.
The Schedule option runs the process as a job. You can track the status of the job from the Home, Jobs tab. This option is best suited for exporting large data sets like All Data or whole chromosome variants. For the scheduling option, you must provide a job name and description.
There is a possibility of replicate and duplicate data in the database. This could be due to loading multiple files belonging to the same specimen_number. This can happen if the same library is sequenced multiple times or the data is reanalyzed. For example, the reads were realigned using the new reference version, so new VCF or gVCF files are created for the same sample. In this scenario, you can use the option to export VCF data only from last loaded files. For example, if variation data has been loaded for a specimen in Jan 2015, Mar 2015 and July 2015, then using this option you can export data from the file loaded in July 2015 and it would not consider variants from the file loaded in Jan and Mar 2015.
Note:
The Schedule Jobs option uses an asynchronous approach to store the file in DBFS. As an alternative to downloading the file using the link in the Oracle Healthcare Translational Research Jobs page, there are other ways to access DBFS. From a Linux OS, you can mount DBFS using dbfs_client application and then browse the directories. Windows OS does not support the FUSE interface and cannot mount DBFS directly. However, there is a dbfs_client application for Windows that can execute commands to access DBFS. The Windows version of dbfs_client lets you use the command line to execute normal directory commands. You can list the DBFS directories as well as copy data from DBFS to the local drive. The dbfs_client application is part of the standard Oracle client software.For more information about using dbfs_client, see http://docs.oracle.com/cd/E11882_01/appdev.112/e18294/adlob_client.htm#ADLOB0006.