23Regulatory Reporting
Regulatory Reporting
About Regulatory Reporting
This chapter describes how to use the Siebel Business Application to create and manage regulatory reports for product complaints and adverse events. The reports are generated with unique and sequential numbers and formatted ready for submission to the FDA or other regulatory agencies.
Where to Find More Information
The following chapters describe other aspects of the complaints and adverse events management process:
About Regulatory Reports
From the time an adverse event or complaint is confirmed as reportable, companies generally have less than 30 days to report to a regulatory agency. An adverse event or complaint can be verified as reportable at various stages of the AECM process, from the initial customer call through to the review of the findings by the analysis team. Companies usually follow certain assessments that determine if an adverse event or complaint is reportable or not.
Separate reports have to be filed for each product that malfunctioned or caused the adverse event. Reports filed are based on the product issue, which typically contains much of the needed information. After the initial report, companies generally file additional supplemental, summary, and annual reports to update information on the investigation and resolution process.
The preconfigured Siebel AECM module supports MedWatch and MDV reports:
-
MedWatch Reports.
Using Siebel AECM, you can automatically generate MedWatch 3500A reports. Sections A to H of the report are populated from the relevant fields in the regulatory report record. The completed report can be saved and submitted electronically to the FDA using the electronic Medical Device Reporting (eMDR) process.
For more information about eMDR refer to About Electronic Medical Device Reporting.
-
MDV Reports.
Medical device companies operating in multiple countries report adverse events or complaints to the various NCAs using the MDV form. Unlike MedWatch reports, there is no standard template for the MDV form. However, the Global Harmonization Task Force (GHTF) has guidelines.
The report created by Siebel AECM follows these guidelines. For more information, see http://www.ghtf.org.
About Report Types
The following table details the four regulatory reports that can be created in Siebel AECM.
Table Report Types in Siebel AECM
| Report Type | Use | Data for the Report Is Taken from These Views in the Regulatory Reports Screen |
|---|---|---|
3500A |
For initial mandatory reports to MedWatch (FDA). |
|
3500A Supplemental |
For additional information to MedWatch after initial reports have been submitted. |
|
MDV Initial |
For initial reports to NCAs who accept MDV forms. |
|
MDV Follow-up |
For additional information to NCAs after initial reports have been submitted. |
|
About Electronic Medical Device Reporting
For customers using the AECM process within the United States, you can submit an eMDR using the Siebel Business Application. Doing so transmits an XML message in HL7 ICSR format to the FDA. Moreover, you can also manage batch submission, and view their confirmation status by referring to the FDA Status field once they have been submitted.
The FDA has created a single system to accept individual, batch, and summary medical device adverse event reports by way of the FDA electronic gateway server. For additional information about the FDA electronic gateway server and FDA standards for eMDR from the FDA, refer to http://www.fda.gov/ForIndustry/FDAeSubmitter/ucm107914.htm.
For information about eMDR transmission using the Siebel Business Application, refer to Transmitting Electronic Medical Device Reporting Immediately, and Managing Batch Electronic Medical Device Reporting.
About FDA Confirmations for Electronic Medical Device Reporting
Once an eMDR or a batch eMDR has been transmitted to the FDA, the FDA Electronic Submissions Gateway (ESG) will respond to the submission. Depending on the processing state, the FDA Status field may be in any of the following three states:
1 of 3 - ESG. Indicates that FDA ESG has sent a Message Delivery Notification (MDN) to confirm that the submission was received. The message includes an official date received.
2 of 3 - ESG. Indicates that FDA ESG has sent an MDN to confirm that the submission has reached the Center for Devices and Radiological Health (CDRH).
3 of 3 - ESG. Indicates that CDRH has sent a message to confirm that the submission has been successfully loaded into the Adverse Event database, or that it has been rejected.
For information about eMDR transmission using Siebel Business Application, refer to Transmitting Electronic Medical Device Reporting Immediately, and Managing Batch Electronic Medical Device Reporting.
Scenario for Regulatory Reporting
This scenario is an example process performed by the Siebel administrator and the quality manager. Your company might follow a different process according to its business requirements.
This scenario is designed to illustrate the functionality of Siebel AECM.
Introduction
A complaint about a cartridge for a blood analyzer machine has been made to the manufacturer. The complaint has been assessed; it needs to be reported to the FDA. A product issue record has already been set up and contains a lot of information about the complaint.
The Siebel Administrator
The administrator is responsible for setting up the report number schema for the organization. The company started using Siebel AECM in the middle of the year. Because the company had already submitted eight MedWatch 3500A reports to the FDA, the administrator sets the report number sequence to start at 9.
Quality Manager
It is the quality manager’s responsibility to prepare the initial MedWatch 3500A form and send it to the FDA. First, she creates a new regulatory report record and populates the record with data from the product issue. Then she reviews the data.
Because her company manufactures the cartridge, she enters data into the Manufacturer and Investigation views. This data will appear in sections G and H of the MedWatch 3500A form.
Satisfied that the necessary data has been entered, she generates the MedWatch 3500A report. This report is a facsimile of the MedWatch 3500A form. But, it is also a standard Siebel report, so the quality manager prints it as she would any other Siebel report.
After reviewing the printed version of the report, she submits it. When she submits the report record, two things happen:
A report number is generated.
Most fields in the report record become read-only.
It is the submitted version of the MedWatch 3500A form that the quality manager sends to the FDA.
The quality manager complied with regulations by sending her initial report within 30 days of the company becoming aware of the issue. However, the product analysis had not been completed at that time. Later, the product analysis team discovers the root cause of the cartridge failure, and the quality manager submits a supplemental report to the FDA.
Process of Regulatory Reporting
This example process represents the tasks that are carried out in the Scenario for Regulatory Reporting.
Additional End-User Procedure
This end-user procedure is not part of the scenario described:
Setting Up Report Numbers
The administrator determines the numbering scheme for regulatory reports. The number is generated automatically when the regulatory report record is submitted. The report number is made up of concatenated fields. The report number field is incremented by 1 each time a report is generated for a new product issue.
| Reports numbers of this type... | Are the concatenation of these fields... | Example |
|---|---|---|
MedWatch 3500A |
|
XERCO-2003-0004 |
MDV Initial |
|
MEDDEV-000008 |
This task is a step in Process of Regulatory Reporting.
To set up report numbering scheme
Navigate to Administration - Group screen, then the Organizations view.
In the Organizations list, select your organization.
To set up numbering for MedWatch 3500A reports, complete the following fields.
Field Comments Manufacturer Id
None.
Report Year
None.
Report Number
Set to zero to have the first report numbered 0001.
To set up numbering for MDV Initial reports, complete the following fields.
Field Comments MDV Id
MDV Report Number
Set to zero to have the first report numbered 000001.
Creating and Populating New Regulatory Reports
The best way to create a regulatory report record is from the product issue record. When a report is created from the regulatory report, fields (for example, products) can be populated from the product issue record to the regulatory report record.
There are two kinds of initial regulatory report that you can create:
MedWatch 3500A
MDV Initial
This task is a step in Process of Regulatory Reporting.
To create a regulatory report record
Navigate to Product Issues screen, then the Product Issue List view.
In the Product Issue List, drill down on a product issue.
Click the Regulatory Reports tab.
In the Regulatory Reports list, create a new record and complete the necessary fields.
When a regulatory report is created:
The following fields are copied from the product issue: PI# and SR #
The following fields are set with a default value: Status is set to In Progress, Owner is set to Creator of the record
For the new record, set the Report Type field to MedWatch 3500A or MDV Initial.
Click Populate Report.
This starts the LS Medical Product Issue Populate Report workflow, which copies data from the product issue according to the report type. For more information about the workflow, see LS Medical Product Issue Populate Report Workflow.
Entering and Reviewing Data for 3500A Reports
When you populate a regulatory report, many fields are copied from the product issue record. When you edit these fields in the regulatory report record, the edits are not copied back to the product issue record.
This task is a step in Process of Regulatory Reporting.
To review and enter data for the 3500A report
Navigate to Regulatory Reports screen, then the Regulatory Report List view.
In the Regulatory Report List, drill down on a 3500A regulatory report record.
Review and if necessary modify the information in the More Info and Patient views.
If you are a manufacturer:
Make sure that the Facility Type field in the Importer view is blank. By default, this field is set to blank.
Complete the fields in the Manufacturer view.
These fields populate section G of the 3500A form. Some fields are described in the following table. Note that the letter and number combination in the last column indicates how this field maps to the 3500A form.
Field Comments Mapping to 3500A Form (A)NDA #
The abbreviated new drug application or the new drug application number.
This field is automatically populated if there is an (A)NDA number associated with the protocol site.
G5
10-day
Select the check box to indicate the report is a 10-day report.
G7
15-day
For reports of serious and unexpected adverse events.
G7
5-day
For events requiring remedial action to prevent unreasonable risk to public health, or where written notice is required.
G7
Address
Manufacturer contact office address - Street.
G1
AE Terms
List of adverse event terms that most accurately characterize the adverse event described in Event Detail section.
G8
City
Manufacturer contact office address - City.
G1
Consumer
Report Source is the consumer or treating health care provider.
G3
Contact Name
Manufacturer contact’s last name.
G1
Contact Office
Manufacturer’s contact office.
G1
Country
Manufacturer contact office address - Country.
G1
Distributor
Check this if report was received from the distributor (importer) of the suspect product.
G3
First Name
Manufacturer contact’s first name.
G1
Follow-up
Check if the report is a follow-up to a previously submitted report.
G7
Follow-Up #
Follow-up sequence number.
G7
Foreign
Report Source is a foreign source (for example, foreign medical facility, affiliate, or government).
G3
IND #
The investigational new drug (IND) application number.
This field is automatically populated if there is an IND number associated with the protocol site.
G5
Initial
Check if the report is the first submission of a manufacturer report (30 day report for device).
G7
Literature
Report Source is the scientific literature or an unpublished manuscript.
G3
Mfg Report #
Regulatory report number.
G9
OTC Product
Check if the suspect medication can be purchased over-the-counter (without a prescription).
G5
Other
Report Source is any source not covered by the previous categories.
G3
Periodic
For reports of serious labeled and non-serious (labeled and unlabeled) adverse events.
G7
Phone #
Manufacturer contact’s work phone number.
G2
PI Received
The date when a company representative became aware of the event.
G4
Postal Code
Manufacturer contact office address - Postal Code.
G1
Pre-1938
Check the box if the suspect medication was marketed prior to 1938 and does not have an NDA #.
G5
Products
Product(s) involved in the event.
Professional
Report Source is a physician, pharmacist, nurse, and so on.
G3
Protocol #
Protocol number identifies the clinical trial at a site. If regulatory report is an IND safety report, enter the protocol number.
G6
Received Report #
Report number for the MedWatch form received from a Importer or a User Facility.
MedWatch Header
Representative
Check this if a company representative reported the event based on information from a health professional.
G3
State
Manufacturer contact office address - State.
G1
Study
Report Source is a postmarketing, clinical trial, surveillance, or other study.
G3
User Facility
Check this if the manufacturer received the report from the MDR contact in a user facility as identified in section F.
G3
If you are a device manufacturer, complete the fields in the Investigation view.
These fields populate section H of the 3500A form. Some fields are described in the following table. Note that the letter and number combination in the last column indicates how this field maps to the 3500A form.
Field Comments Mapping to 3500A Form Evaluation
If an evaluation was conducted, note summary here and choose Evaluation Summary Attached in Evaluated by Mfg Field.
H3
Death
Check only if the death was an outcome of the adverse event.
H1
Correction
Do not check when creating an initial report.
Follow-up with changes to previously submitted information.
H2
Serious Injury
Event is life-threatening, results in permanent impairment, requires intervention to prevent permanent impairment.
H1
Additional Information
Do not check when creating an initial report.
Information concerning the event that was not provided in the initial report.
H2
Malfunction
Device malfunctions.
H1
Response to FDA Request
Do not check when creating an initial report.
Additional information requested by FDA concerning the device/event.
H2
Other
Event not covered by death, serious injury, or malfunction. This type of category should be rarely used.
H1
Device Evaluation
Do not check when creating an initial report.
Evaluation/analysis of device.
H2
Mfg Narrative
Any additional information, evaluation, or clarification of data presented in previous sections.
H10
Recall
Remedial Action - Recall.
H7
Method Codes
Method codes capture two items — the source of the device that was evaluated and the type of evaluation performed.
Do not enter more than four codes.
H6
Repair
Remedial Action - Repair.
H7
Result Codes
Describes the results of evaluation and analyses of the reported device problem(s).
Do not enter more than four codes.
H6
Replace
Remedial Action - Replace.
H7
Conclusion Codes
Describes the evaluation conclusions.
Do not enter more than four codes.
H6
Relabeling
Remedial Action - Relabeling.
H7
Evaluated by Mfg
If you do not check this box, then you should complete the Non-Evaluation Codes field.
Identify if the device was evaluated.
H3
Notification
Remedial Action - Notification.
H7
Non-Evaluation Codes
If an evaluation of a returned medical device was NOT conducted, provide the appropriate code.
H3
Corrected Data
Additional, corrected, or missing information, identifying each data item by the applicable section and block number.
H11
Inspection
Remedial Action - Inspection.
H7
Usage of Device
Indicates whether the use of the suspect medical device was the initial use, reuse, or unknown.
H8
Patient Monitoring
Remedial Action - Patient monitoring.
H7
Mfg Date
Month and year of manufacture of the suspect medical device.
This field can be based on asset number (asset manufacture date) or lot number (effective start date).
H4
Modification
Remedial Action - Modification.
H7
Labeled Single Use
Indicates whether the device was labeled for single use.
H5
Other
Remedial Action - Other - Specify the type of action in this field.
H7
Correction #
If action reported to FDA under 21 USC 360i(f), list correction or removal reporting number.
H9
If you are a user facility or importer, complete the fields in the Importer view.
These fields populate section F of the form. Some fields are described in the following table. Note that the letter and number combination in the last column indicates how this field maps to the 3500A form.
Field Comments Mapping to 3500A Form Facility Type
Indicate whether the report is from a user facility, importer, or others.
F1
Importer
Name of the distributor or importer.
F3
Contact Name
Last name of the distributor’s or importer’s representative to contact regarding the event.
F4
Device Age
The approximate age of the device.
F9
Report #
Regulatory report # for this report, which is being submitted by an importer.
This number is auto-populated when the report is generated.
F2
Address
Distributor’s or Importer’s address - Street Address line #1.
This field is auto-populated based on the distributor’s or importer’s name.
F3
First Name
First name of the distributor’s or importer’s representative to contact regarding the event.
F4
Age UoM
Unit of measurement for device age.
F9
Report Type
Indicates if the report to the regulatory agency will be an initial or follow-up report.
F7
City
Distributor’s or importer’s address - City.
F3
Phone #
Contact’s work phone number.
F5
Patient Codes
Patient codes describe what happened to the patient as a result of the event.
Do not enter more than three codes.
F10
Follow-up #
Sequence number of the follow-up report.
F7
Postal Code
Distributor’s or importer’s address - Postal Code.
F3
Reported FDA
Indicates if the distributor or importer has already sent a report to the regulatory agency.
F11
Device Codes
Device codes describe device failures or problems encountered during the event.
Do not enter more than four codes.
F10
Reported Mfg
Indicates if the distributor or importer has sent a report to the manufacturer.
F13
State
Distributor’s or importer’s address - State.
F3
FDA Report Date
Date the report was sent to the regulator agency.
F11
Event Location
Location of the actual occurrence of the event.
F12
Mfg Report Date
Date the report was sent to the manufacturer.
F13
Country
Distributor’s or importer’s address - Country.
F3
PI Received
The date when a company representative became aware of the event.
F6
Entering and Reviewing Data for MDV Reports
When you populate the regulatory report, many fields are copied from the product issue record. When you edit these fields in the regulatory report record, the edits are not copied back to the product issue record.
This task is a step in Process of Regulatory Reporting.
To review and enter data for an MDV Initial report
Navigate to Regulatory Reports screen, then the Regulatory Report List view.
In the Regulatory Report List, drill down on an MDV regulatory report record.
Review and if necessary modify the information in the More Info and Patient views.
Click the MDV tab.
On the MDV form, complete the necessary fields.
Some fields are described in the following table.
Field Comments MDV Report #
MDV Report number.
This number is auto-populated when the report is generated.
Authorities
The name of the regulatory agencies to send the report to.
Affiliate Acct
The affiliate account representing the company.
PI Received
The date when the company became aware of the event.
Determination
The type of the reportable event for MDV reporting.
Patient Codes
Patient codes describe what happened to the patient as a result of the event.
Device Codes
Device codes describe device failures or problems encountered during the event.
Usage of Device
Indicates whether the use of the suspect medical device was the initial use, reuse, or unknown.
Follow-up Date
Date field to indicate the approximate follow-up or final report date.
Approval Org
The approval body for the device.
Approval #
The approval number for the device.
Evaluation
If the evaluation was conducted, note summary here and choose Evaluation Summary Attached in Evaluated by Mfg field.
Mfg Narrative
Any additional information, evaluation, or clarification of data presented in previous sections.
Running 3500A and MDV Reports
Siebel AECM uses the Siebel Reports Server to create formatted reports suitable for submitting to the FDA or other regulatory agency.
Typically, the user views the report:
Before generating it, to make sure that the data is correct
After generating it, to print a final report for sending to the FDA or other regulatory agencies
This task is a step in Process of Regulatory Reporting.
To run a report
Navigate to the Regulatory Reports screen, then the Regulatory Report List view.
In the Regulatory Report List, drill down on a report.
For this type of report... Navigate to one of these views... Select this report from the Reports button menu MedWatch 3500A
More Info
- Patient
- Importer
- Manufacturer
- Investigation
3500A
MVD
More Info
- Patient
- MDV
MDV
Run, print, and save the report as necessary. For information about reports, see Siebel Fundamentals.
Which Sections of the Med Watch 3500A Form Are Filled In?
Not all sections of the MedWatch 3500A are filled in for every report. Which sections are filled in depends upon the Event Type and Facility Type fields. The Event Type field is in the More Info view of the Regulatory Reports screen. The Facility Type field is in the Importer view of the Regulatory Reports screen.
These sections always are filled in:
A. Patient Information
B. Adverse Event or Product Problem
E. Initial Reporter
The following information lists the conditions for which the other sections get filled in.
Table Sections of the Report Filled In According to Event Type and Facility Type Field Values
If... And... These sections are filled in... And these sections can remain blank... Event Type = Adverse Event Drug, Product Problem Drug, or AE and PP Drug
—
C
G
D
F
H
Event Type = Adverse Event Device, Product Problem Device, or AE and PP Device
Facility Type = NULL (blank)
D
G
H
C
F
Event Type = Adverse Event Device, Product Problem Device, or AE and PP Device
Facility Type = User Facility, Distributor, or Importer (That is, not blank)
D
F
G
C
H
Generating Regulatory Report Numbers and Submitting Reports
When you generate a 3500A or MDV initial report:
The report number is generated and filled in
The Status field changes to Submitted
The fields in the regulatory report record become read-only (except for the Sub Status field)
Only the primary owner of the regulatory report can generate the report number and submit the report.
This task is a step in Process of Regulatory Reporting.
To generate and submit a regulatory report
Make sure the application is running on a server database.
You cannot generate a report number on the Siebel Mobile Web Client.
Navigate to Regulatory Reports screen, then the My Regulatory Reports view.
In the Regulatory Report List, drill down on a regulatory report with status of In Progress.
Click Generate.
This starts the LS Medical Product Issue RR Submit workflow, which authenticates the user, adds a number for the regulatory report, changes the status to Submitted, and makes all fields except Sub Status read-only.
For more information about the workflow, see LS Medical Product Issue RR Submit Workflow.
To submit the regulatory report to the FDA, click eMDR Queue.
This changes the Sub Status of the report to eMDR and places the report in the queue to be sent to the FDA at the end of the month. If you want to submit the report immediately, follow the instructions outlined in Transmitting Electronic Medical Device Reporting Immediately.
Transmitting Electronic Medical Device Reporting Immediately
Occasionally, you may be made aware of a unique medical device report that cannot wait until month-end processing, and must be reported to the FDA immediately.
For more information about rules and guidelines for regulatory reporting delays, refer http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm149672.htm.
To force immediate FDA reporting
Navigate to Regulatory Reports screen, then the eMDR view.
A list of all regulatory reports whose RR Sub Status is eMDR will be listed.
In the eMDR list, select the regulatory report you want to transmit.
Regulatory reports that have not yet been transmitted will have Transmission Status set to Not Transmitted.
Click Transmit.
Note: The Transmit button will be grayed out if the regulatory report has already been transmitted or has been overridden.
Once transmitted, monitor the FDA Status field of the eMDR for FDA confirmation. For more information on confirmation statuses, refer to About FDA Confirmations for Electronic Medical Device Reporting.
Managing Batch Electronic Medical Device Reporting
Occasionally, you may want to submit multiple medical device reports as a batch. For example, if you are submitting 100 month-end reports, creating a batch enables you to submit reports in one transmission rather than 100 individual transmissions.
To create a batch e MDR and populate it with regulatory reports
Navigate to Regulatory Reports screen, then the eMDR Batch view.
A list of existing eMDR batches will appear.
In the eMDR batch list, create a new record and complete the necessary fields.
For the new record, enter any unique alpha numeric string in the Batch # field.
Navigate to Regulatory Reports screen, then the eMDR view and do the following:
Identify a regulatory report that you want to include in batch, and modify the Batch # field to the one specified in Step 3.
Repeat Step a for every regulatory report that you want to include in the batch.
This regulatory reports will remain in the eMDR queue and will be sent as part of a batch to the FDA at month end.
To transmit the batch immediately, do the following:
Navigate to Regulatory Reports screen, then the eMDR Batch view,
Select the batch created in Step 3, and then click Transmit.
Reopening a Regulatory Report
If you want to make a change in a generated (submitted) regulatory report record, you need to reopen it. Only the primary owner of the regulatory report can reopen it.
When you reopen a report:
The Status field changes to Reopen
The fields in the regulatory report record can be edited
This task is a step in Process of Regulatory Reporting.
To reopen a regulatory report
Navigate to Regulatory Reports screen, then the Regulatory Report List view.
In the Regulatory Report List, drill down on a regulatory report with status of Submitted.
Click Reopen.
This starts the LS Medical Product Issue RR Reopen workflow, which authenticates the user and changes the status of the report to Reopen.
For more information about the workflow, see LS Medical Product Issue RR Reopen Workflow.
Creating Supplemental or Follow-Up Regulatory Reports
Often, it is necessary to follow-up an initial report with a follow-up report that provides supplementary information to the regulatory agency.
When you run a supplemental or follow-up report, the report should contain the initial report number and any corrected or new data.
This task is a step in Process of Regulatory Reporting.
To create a follow-up or supplemental regulatory report
Navigate to Product Issues screen, then the Product Issue List view.
In the Product Issue List, drill down on a product issue.
Click the Regulatory Reports tab.
In the Regulatory Reports list, create a new record and complete the necessary fields.
When the regulatory report is created:
The following fields are copied from the product issue: PI # and SR #
The following fields are set with a default value: Status is set to In Progress, Owner is set to Creator of the record.
Set the Report Type field to 3500A Supplemental or MDV Follow-up.
Click Populate Report.
The Populate Report button only copies the initial report number to the supplemental report. (No other fields are copied.) You can configure the application to copy additional fields such as Mfg Narrative and Corrected Data to the supplemental report. Review the LS Medical Product Issue Populate Report Workflow for an example of a similar configuration.
If you are a device manufacturer preparing a 3500A Supplemental report, make sure to check a Follow-Up Type in the Investigation view.
After creating a follow-up or supplemental regulatory report, you can view it, submit it, reopen it in the same way as a initial report:
Field Mapping for the MedWatch Report
The following table shows how the fields in the Regulatory Reports screen map to the fields in the MedWatch 3500A report. If the content for text fields extends to continuation pages, it starts from the third page. Note the following about the different areas on the Location MedWatch Form 3500A:
A: Patient Information
B: Adverse Event or Product Problem
C: Suspect Medication(s)
D: Suspect Medical Device
E: Initial Reporter
F: For Use by User Facility/Importer (Devices Only)
G: All Manufacturers
H: Device Manufacturers Only
Table Mapping of Fields from the Siebel AECM UI to the Med Watch 3500A Form
Location on MedWatch Form 3500A Field Name View (Applet) on the Regulatory Reports and the Product Issues screen A1
Patient Identifier
Patient
A2
Age
Patient
A2
Date of Birth
Patient
A3
Gender
Patient
A4
U/M
Patient
A4
Weight
Patient
B1
Event Type
More Info (Event Detail)
B2
Congenital Anomaly
More Info (Event Detail)
B2
Date of Death
More Info (Event Detail)
B2
Death
More Info (Event Detail)
B2
Disability
More Info (Event Detail)
B2
Hospitalization
More Info (Event Detail)
B2
Life Threatening
More Info (Event Detail)
B2
Other
More Info (Event Detail)
B2
Required Intervention
More Info (Event Detail)
B3
Event Date
More Info (Event Detail)
B4
Report Date
More Info, Regulatory Reports screen only
B5
Description
More Info (Event Detail)
B6
Tests/Data
More Info (Event Detail)
B7
Relevant History
More Info (Event Detail)
C1
Product
More Info (Products)
C2
Dose Per Unit
More Info (Products)
C2
Frequency
More Info (Products)
C2
Route Used
More Info (Products)
C3
Therapy From Date
More Info (Products)
C3
Therapy To Date
More Info (Products)
C4
Indication
More Info (Products)
C5
Event Abated
More Info (Products)
C6
Lot #
More Info (Products)
C7
Expiration Date
More Info (Products)
C8
Reintroduce Reoccur
More Info (Products)
C9
NDC#
More Info (Products)
C10
External Products
More Info (Event Detail)
D1
Product
More Info (Products)
D2
Common Device Name
More Info (Products)
D3
City
More Info (Products)
D3
Mfg Name
More Info (Products)
D3
Postal Code
More Info (Products)
D3
State
More Info (Products)
D3
Street Address
More Info (Products)
D4
Asset #
More Info (Products)
D4
Catalog #
More Info (Products)
D4
Expiration Date
More Info (Products)
D4
Lot #
More Info (Products)
D4
Model #
More Info (Products)
D4
Part #
More Info (Products)
D4
Serial #
More Info (Products)
D5
Device Operator
More Info (Products)
D6
Implant Date
More Info (Products)
D7
Explant Date
More Info (Products)
D8
Reprocessed
More Info (Products)
D9
Reprocessor
More Info (Products)
D10
Device Available
More Info (Products)
D10
Return Date
More Info (Products)
D11
External Products
More Info (Event Detail)
E1
Account
More Info
E1
Address
More Info
E1
City
More Info
E1
Contacts (Contact Last Name in list)
More Info
E1
CSN #
More Info
E1
First Name (Contact First Name in list)
More Info
E1
Phone #
More Info
E1
Postal Code
More Info
E1
Site
More Info
E1
State
More Info
E2
Provider
More Info
E3
Occupation
More Info
E4
Reported FDA
More Info
F1
Facility Type
Importer
F2
Report #
Importer
F3
Address
Importer
F3
City
Importer
F3
Country
Importer
F3
Importer
Importer
F3
Postal Code
Importer
F3
State
Importer
F4
Contact Name
Importer
F4
First Name
Importer
F5
Phone #
Importer
F6
PI Received
Importer
F7
Follow-up #
Importer
F7
Report Type
Importer
F8
Report Date
More Info
F9
Age UoM
Importer
F9
Device Age
Importer
F10
Device Codes
Importer
F10
Patient Codes
Importer
F11
FDA Report Date
Importer
F11
Reported FDA
Importer
F12
Event Location
Importer
F13
Mfg Report Date
Importer
F13
Reported Mfg
Importer
G1
Address
Manufacturer
G1
City
Manufacturer
G1
Contact Name
Manufacturer
G1
Contact Office
Manufacturer
G1
Country
Manufacturer
G1
First Name
Manufacturer
G1
Postal Code
Manufacturer
G1
State
Manufacturer
G2
Phone #
Manufacturer
G3
Consumer
Manufacturer
G3
Foreign
Manufacturer
G3
Distributor
Manufacturer
G3
Literature
Manufacturer
G3
Other
Manufacturer
G3
Professional
Manufacturer
G3
Representative
Manufacturer
G3
Study
Manufacturer
G3
User Facility
Manufacturer
G4
PI Received
Manufacturer
G5
(A)NDA #
Manufacturer
G5
IND #
Manufacturer
G5
OTC Product
Manufacturer
G5
Pre-1938
Manufacturer
G6
Protocol #
More Info (Product Issues) and Manufacturer
G7
10-day
Manufacturer
G7
15-day
Manufacturer
G7
5-day
Manufacturer
G7
Follow-up
Manufacturer
G7
Follow-Up #
Manufacturer
G7
Initial
Manufacturer
G7
Periodic
Manufacturer
G8
AE Terms
Manufacturer
G9
Mfg Report #
Manufacturer
H1
Death
Investigation
H1
Malfunction
Investigation
H1
Other
Investigation
H1
Serious Injury
Investigation
H2
Additional Information
Investigation
H2
Correction
Investigation
H2
Device Evaluation
Investigation
H2
Response to FDA Request
Investigation
H3
Evaluated by Mfg
Investigation
H3
Evaluation
Investigation
MDV
H3
Non-Evaluation Codes
Investigation
H4
Mfg Date
Investigation
More Info (Products)
H5
Labeled Single Use
More Info (Products)
Investigation
H6
Conclusion Codes
Investigation
H6
Method Codes
Investigation
H6
Result Codes
Investigation
H7
Inspection
Investigation
H7
Modification
Investigation
H7
Notification
Investigation
H7
Other
Investigation
H7
Patient Monitoring
Investigation
H7
Recall
Investigation
H7
Relabeling
Investigation
H7
Repair
Investigation
H7
Replace
Investigation
H8
Usage of Device
Investigation
H9
Correction #
Investigation
H10
Mfg Narrative
Investigation
MDV
H11
Corrected Data
Investigation
Field Mapping for the MDV Report
The following table shows how the fields in the Regulatory Reports screen map to the fields in the MDV report.
Table Mapping of Fields from Siebel AECM UI to the MDV Report
| Regulatory Reports Screen | UI Field Name | View (Applet) on the Regulatory Reports and the Product Issues screen | Text on MDV Report |
|---|---|---|---|
Report Header |
Report # MDV Report # |
More Info MDV |
MDV Report # |
Report Header |
Affiliate Ref # |
MDV |
Affiliate Ref # |
Report Header |
Type |
More Info |
Report Type |
MDV Determination |
Determination |
MDV |
MDV Determination |
Destination |
Authorities |
MDV |
Competent Authority Name |
Destination |
Address Address 2 City State Postal Code Country |
MDV |
Address |
General |
Report Date |
More Info, Regulatory Reports screen only |
Report Date |
General |
Received |
More Info |
Manufacturer Awareness Date |
General |
Affiliate Acct |
MDV |
Reporting Firm Name |
General |
Auth Rep First Name |
MDV |
Authorized Representative |
General |
Address City State Postal Code Country |
MDV |
Address |
General |
Phone # |
MDV |
Telephone |
General |
Fax # |
MDV |
Facsimile |
Event Information |
Patient Identifier |
Patient |
Patient Identifier |
Event Information |
Gender |
Patient |
Gender |
Event Information |
Age |
Patient |
Age |
Event Information |
Weight |
Patient |
Weight |
Event Information |
Date of Birth |
Patient |
Date of Birth |
Event Information |
Account |
More Info |
Health Care Facility Name |
Event Information |
Site |
More Info |
Site |
Event Information |
Contacts, First Name |
More Info |
Contact Name |
Event Information |
Address |
More Info |
Address |
Event Information |
Phone # |
More Info |
Contact Phone Number |
Event Information |
Event Date |
More Info (Event Detail) |
Event Date |
Event Information |
Description |
More Info (Event Detail) |
Event Description |
Event Information |
# Occurrences |
More Info (Event Detail) |
Number of Occurrences |
Event Information |
Labeled Single Use |
More Info (Products) Investigation |
Device Labeled for Single Use |
Event Information |
Patient Codes |
MDV Importer |
Patient Outcomes |
Event Information |
Device Codes |
MDV Importer |
Device Outcomes |
Event Information |
Usage of Device |
MDV Investigation |
Usage of Device |
Event Information |
Authorities |
MDV |
Competent Authorities Already Notified |
Device Information |
Mfg Name |
MDV |
Manufacturer Name |
Device Information |
Contact First Name |
MDV |
Manufacturer Contact Name |
Device Information |
Address City State Postal Code Country |
MDV |
Address |
Device Information |
Phone # |
MDV |
Telephone |
Device Information |
Fax # |
MDV |
Facsimile |
Device Information |
Product |
More Info (Products) |
Device Name |
Device Information |
Device Type |
More Info (Products) |
Type of Device |
Device Information |
Lot # |
More Info (Products) |
Device Lot # |
Device Information |
Part # |
More Info (Products) |
Device Part # |
Device Information |
Asset # |
More Info (Products) |
Device Asset # |
Device Information |
Serial # |
More Info (Products) |
Device Serial # |
Device Information |
Model # |
More Info (Products) |
Model # |
Device Information |
Catalog # |
More Info (Products) |
Catalog # |
Device Information |
Approval Org |
MDV |
Device Approval Body |
Device Information |
Approval # |
MDV |
Approval # |
Device Information |
Qty Involved |
More Info (Products) |
Quantity of Devices Involved |
Device Information |
Device Available |
More Info (Products) |
Device Disposition |
Device Information |
External Products |
More Info (Event Detail) |
Associated Products |
Results of Investigation |
Evaluation |
MDV Investigation |
Device Evaluation Result |
Results of Investigation |
Remedials |
MDV |
Remedial Action |
Results of Investigation |
MFG Narrative |
MDV Investigation |
Manufacturer’s Narrative |
About Configuring Buttons in Regulatory Reports
There are three buttons associated with regulatory reports. You can configure the workflows for these buttons.
Populate Report button (LS Medical Product Issue Populate Report Workflow)
Generate (LS Medical Product Issue RR Submit Workflow)
You can modify workflows to suit your own business model using Siebel Business Process Designer. For more information, see Siebel Business Process Framework: Workflow Guide.
LS Medical Product Issue Populate Report Workflow
This workflow is initiated from the Populate button on the Regulatory Reports view of the Product Issues screen.
The workflow appears in the following figure.
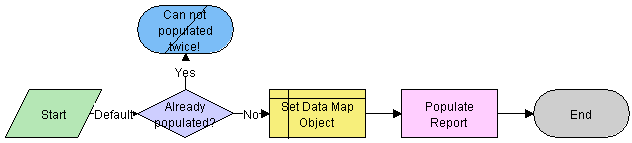
Workflow Description
This workflow performs the following actions:
Checks if current record has already been populated. If it has, the workflow ends.
Sets the data map object according to the report type.
The data map objects are:
LS Medical PI Populate Report - 3500A
LS Medical PI Populate Report - 3500 Supplemental
LS Medical PI Populate Report - MDV Initial
LS Medical PI Populate Report - MDV Follow-up
Populates the report from the product issue according to the data map object.
LS Medical Product Issue RR Submit Workflow
This workflow is initiated from the Generate button on the Regulatory Reports screen.
The workflow appears in the following figure.
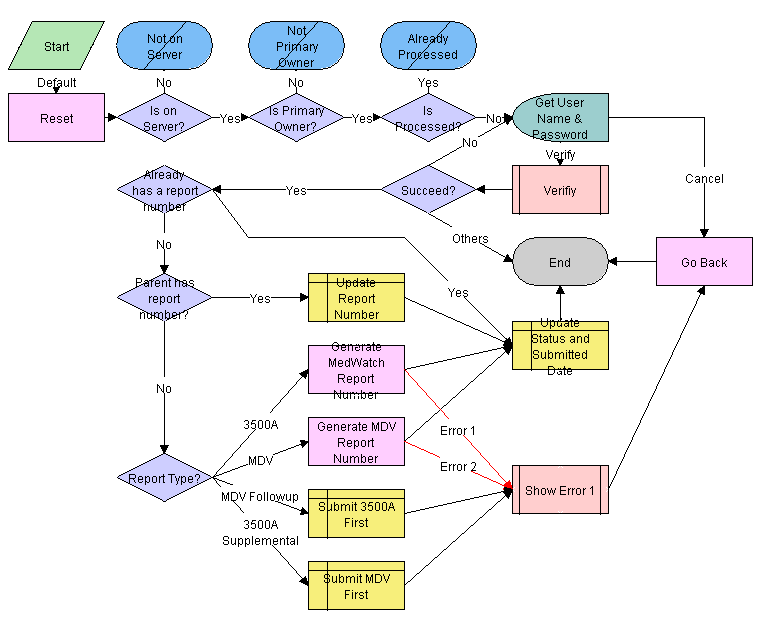
Workflow Description
This workflow performs the following actions:
Checks if the application is running on a server database. If not, the workflow ends.
Checks if the user is the primary owner. If not, the workflow ends.
Checks if the record has already been processed. If it has, the workflow ends.
Calls the LS Medical User Verification workflow. If the authentication does not pass, the workflow ends.
Checks if the parent product issue already has a report number.
If the product issue. . . and the Report Type Is. . . Then. . . Has a report record with a report number
—
Use the same number for the report being submitted.
Does not have a report record with a report number
3500A or MDV
Generate a new report number.
3500A Supplemental or MDV Follow-up
The workflow ends.
Sets the status of the report to Submitted.
LS Medical Product Issue RR Reopen Workflow
This workflow is initiated from the Reopen button on the Regulatory Reports screen.
The workflow appears in the following figure.
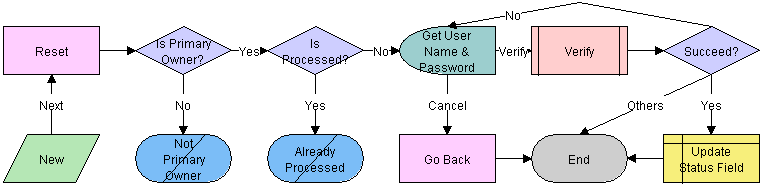
Workflow Description
This workflow performs the following actions:
Checks if the user is the primary owner. If not, the workflow ends.
Checks if the record has already been processed. If it has, the workflow ends.
Calls the LS Medical User Verification workflow. If the authentication does not pass, the workflow ends.
Sets the status of the report to Reopen.